 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Alfredo Gonzalez-Alcocer | -- | 2544 | 2023-01-18 18:12:00 | | | |
| 2 | Alfredo Gonzalez-Alcocer | -102 word(s) | 2684 | 2023-01-18 21:04:56 | | | | |
| 3 | Jason Zhu | Meta information modification | 2684 | 2023-01-19 02:41:51 | | | | |
| 4 | Jason Zhu | + 2 word(s) | 2686 | 2023-02-02 02:16:40 | | |
Video Upload Options
Biometals are all metal ions that are essential for all living organisms. About 40% of all enzymes with known structures require biometals to function correctly. The main target of damage by biometals is the central nervous system (CNS). Biometal dysregulation (metal deficiency or overload) is related to pathological processes. Chronic occupational and environmental exposure to biometals, including iron and copper, is related to an increased risk of developing Parkinson’s disease (PD). Indeed, biometals have been shown to induce a dopaminergic neuronal loss in the substantia nigra. Although the etiology of PD is still unknown, oxidative stress dysregulation, mitochondrial dysfunction, and inhibition of both the ubiquitin–proteasome system (UPS) and autophagy are related to dopaminergic neuronal death.
1. Metals, Parkinson’s Disease, and Oxidative Stress
2. Redox-Active Metals’ Role in Dopaminergic Neuronal Death
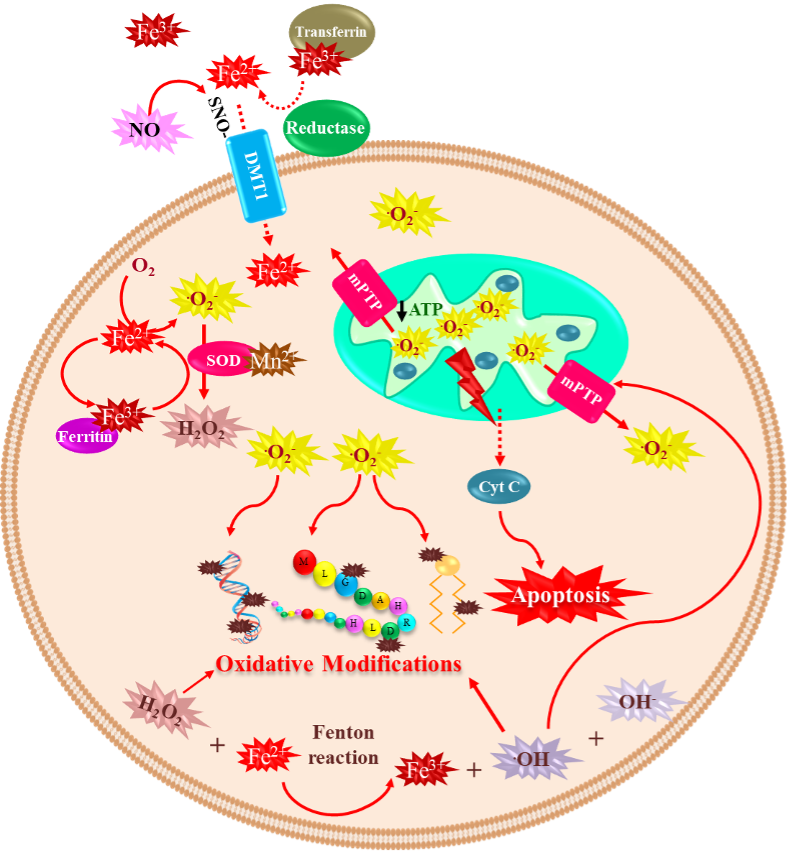
2.1. Iron
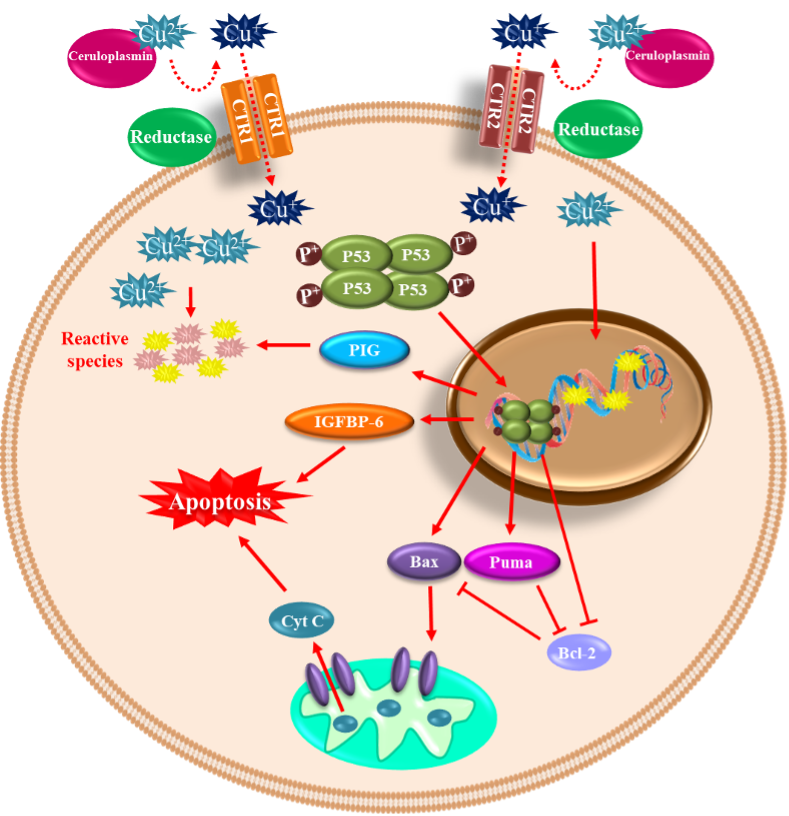
2.2. Copper
References
- McCord, J.M. The evolution of free radicals and oxidative stress. Am. J. Med. 2000, 108, 652–659.
- Martemucci, G.; Costagliola, C.; Mariano, M.; D’Andrea, L.; Napolitano, P.; D’Alessandro, A.G. Free Radical Properties, Source and Targets, Antioxidant Consumption and Health. Oxygen 2022, 2, 48–78.
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative Stress: Harms and Benefits for Human Health. Oxidative Med. Cell. Longev. 2017, 2017, 8416763.
- Gilgun-Sherki, Y.; Melamed, E.; Offen, D. Oxidative stress induced-neurodegenerative diseases: The need for antioxidants that penetrate the blood brain barrier. Neuropharmacology 2001, 40, 959–975.
- Navarro-Yepes, J.; Burns, M.; Anandhan, A.; Khalimonchuk, O.; del Razo, L.M.; Quintanilla-Vega, B.; Pappa, A.; Panayiotidis, M.; Franco, R. Oxidative stress, redox signaling, and autophagy: Cell death versus survival. Antioxid. Redox Signal. 2014, 21, 66–85.
- Kurutas, E.B. The importance of antioxidants which play the role in cellular response against oxidative/nitrosative stress: Current state. Nutr. J. 2016, 15, 71.
- Forman, H.J.; Zhang, H. Targeting oxidative stress in disease: Promise and limitations of antioxidant therapy. Nat. Rev. Drug Discov. 2021, 20, 689–709.
- Stykel, M.G.; Ryan, S.D. Nitrosative stress in Parkinson’s disease. NPJ Parkinsons Dis. 2022, 8, 104.
- Hartmann, A. Postmortem studies in Parkinson’s disease. Dialogues Clin. Neurosci. 2004, 6, 281–293.
- Farzam, A.; Chohan, K.; Strmiskova, M.; Hewitt, S.J.; Park, D.S.; Pezacki, J.P.; Özcelik, D. A functionalized hydroxydopamine quinone links thiol modification to neuronal cell death. Redox Biol. 2020, 28, 101377.
- Cobley, J.N.; Fiorello, M.L.; Bailey, D.M. 13 reasons why the brain is susceptible to oxidative stress. Redox Biol. 2018, 15, 490–503.
- Kish, S.J.; Morito, C.; Hornykiewicz, O. Glutathione peroxidase activity in Parkinson’s disease brain. Neurosci. Lett. 1985, 58, 343–346.
- Chen, C.; Turnbull, D.M.; Reeve, A.K. Mitochondrial Dysfunction in Parkinson’s Disease-Cause or Consequence? Biology 2019, 8, 38.
- Ball, N.; Teo, W.-P.; Chandra, S.; Chapman, J. Parkinson’s Disease and the Environment. Front. Neurol. 2019, 10, 218.
- Dexter, D.T.; Wells, F.R.; Lees, A.J.; Agid, F.; Agid, Y.; Jenner, P.; Marsden, C.D. Increased nigral iron content and alterations in other metal ions occurring in brain in Parkinson’s disease. J. Neurochem. 1989, 52, 1830–1836.
- Nakamura, K.; Bindokas, V.P.; Marks, J.D.; Wright, D.A.; Frim, D.M.; Miller, R.J.; Kang, U.J. The selective toxicity of 1-methyl-4-phenylpyridinium to dopaminergic neurons: The role of mitochondrial complex I and reactive oxygen species revisited. Mol. Pharm. 2000, 58, 271–278.
- Obata, T. Dopamine efflux by MPTP and hydroxyl radical generation. J. Neural Transm. 2002, 109, 1159–1180.
- Hoffman, C.; Aballay, A. Role of neurons in the control of immune defense. Curr. Opin. Immunol. 2019, 60, 30–36.
- Lesage, S.; Brice, A. Parkinson’s disease: From monogenic forms to genetic susceptibility factors. Hum. Mol. Genet. 2009, 18, R48–R59.
- Shults, C.W. Lewy bodies. Proc. Natl. Acad. Sci. USA 2006, 103, 1661–1668.
- Uversky, V.N.; Li, J.; Fink, A.L. Metal-triggered structural transformations, aggregation, and fibrillation of human alpha-synuclein. A possible molecular NK between Parkinson’s disease and heavy metal exposure. J. Biol. Chem. 2001, 276, 44284–44296.
- Liu, L.L.; Franz, K.J. Phosphorylation-dependent metal binding by alpha-synuclein peptide fragments. J. Biol. Inorg. Chem. 2007, 12, 234–247.
- Liu, L.L.; Franz, K.J. Phosphorylation of an alpha-synuclein peptide fragment enhances metal binding. J. Am. Chem. Soc. 2005, 127, 9662–9663.
- Garcia-Garcia, A.; Rodriguez-Rocha, H.; Madayiputhiya, N.; Pappa, A.; Panayiotidis, M.I.; Franco, R. Biomarkers of protein oxidation in human disease. Curr. Mol. Med. 2012, 12, 681–697.
- Dlouhy, A.C.; Outten, C.E. The iron metallome in eukaryotic organisms. Met. Ions Life Sci. 2013, 12, 241–278.
- Casey, J.L.; Hentze, M.W.; Koeller, D.M.; Caughman, S.W.; Rouault, T.A.; Klausner, R.D.; Harford, J.B. Iron-responsive elements: Regulatory RNA sequences that control mRNA levels and translation. Science 1988, 240, 924–928.
- Hentze, M.W.; Muckenthaler, M.U.; Andrews, N.C. Balancing acts: Molecular control of mammalian iron metabolism. Cell 2004, 117, 285–297.
- Arosio, P.; Elia, L.; Poli, M. Ferritin, cellular iron storage and regulation. IUBMB Life 2017, 69, 414–422.
- Bresgen, N.; Eckl, P.M. Oxidative stress and the homeodynamics of iron metabolism. Biomolecules 2015, 5, 808–847.
- Siah, C.W.; Ombiga, J.; Adams, L.A.; Trinder, D.; Olynyk, J.K. Normal iron metabolism and the pathophysiology of iron overload disorders. Clin. Biochem. Rev. 2006, 27, 5–16.
- Núñez, M.T.; Urrutia, P.; Mena, N.; Aguirre, P.; Tapia, V.; Salazar, J. Iron toxicity in neurodegeneration. Biometals 2012, 25, 761–776.
- Cotruvo, J.J.A.; Stubbe, J. Metallation and mismetallation of iron and manganese proteins in vitro and in vivo: The class I ribonucleotide reductases as a case study. Metallomics 2012, 4, 1020–1036.
- Zhu, W.; Li, X.; Xie, W.; Luo, F.; Kaur, D.; Andersen, J.K.; Jankovic, J.; Le, W. Genetic iron chelation protects against proteasome inhibition-induced dopamine neuron degeneration. Neurobiol. Dis. 2010, 37, 307–313.
- Liu, C.; Zhang, C.-W.; Lo, S.Q.; Ang, S.T.; Chew, K.C.M.; Yu, D.; Chai, B.H.; Tan, B.; Tsang, F.; Tai, Y.K.; et al. S-Nitrosylation of Divalent Metal Transporter 1 Enhances Iron Uptake to Mediate Loss of Dopaminergic Neurons and Motoric Deficit. J. Neurosci. 2018, 38, 8364–8377.
- Yarjanli, Z.; Ghaedi, K.; Esmaeili, A.; Rahgozar, S.; Zarrabi, A. Iron oxide nanoparticles may damage to the neural tissue through iron accumulation, oxidative stress, and protein aggregation. BMC Neurosci. 2017, 18, 51.
- Ramos, P.; Santos, A.; Pinto, N.R.; Mendes, R.; Magalhães, T.; Almeida, A. Iron levels in the human brain: A post-mortem study of anatomical region differences and age-related changes. J. Trace Elem. Med. Biol. 2014, 28, 13–17.
- Linert, W.; Jameson, G.N. Redox reactions of neurotransmitters possibly involved in the progression of Parkinson’s Disease. J. Inorg. Biochem. 2000, 79, 319–326.
- Youdim, M.B.; Riederer, P.F. A review of the mechanisms and role of monoamine oxidase inhibitors in Parkinson’s disease. Neurology 2004, 63 (Suppl. 2), S32-5.
- Lu, H.; Chen, J.; Huang, H.; Zhou, M.; Zhu, Q.; Yao, S.Q.; Chai, Z.; Hu, Y. Iron modulates the activity of monoamine oxidase B in SH-SY5Y cells. Biometals 2017, 30, 599–607.
- Napolitano, A.; Manini, P.; d’Ischia, M. Oxidation chemistry of catecholamines and neuronal degeneration: An update. Curr. Med. Chem. 2011, 18, 1832–1845.
- Barbusinski, K. Toxicity of Industrial Wastewater Treated by Fenton’s Reagent. Pol. J. Environ. Stud. 2005, 14, 11–16.
- Kehrer, J.P. The Haber-Weiss reaction and mechanisms of toxicity. Toxicology 2000, 149, 43–50.
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950.
- Gilman, C.P.; Chan, S.L.; Guo, Z.; Zhu, X.; Greig, N.; Mattson, M.P. p53 is present in synapses where it mediates mitochondrial dysfunction and synaptic degeneration in response to DNA damage, and oxidative and excitotoxic insults. Neuromolecular Med. 2003, 3, 159–172.
- Koskenkorva-Frank, T.S.; Weiss, G.; Koppenol, W.H.; Burckhardt, S. The complex interplay of iron metabolism, reactive oxygen species, and reactive nitrogen species: Insights into the potential of various iron therapies to induce oxidative and nitrosative stress. Free Radic. Biol. Med. 2013, 65, 1174–1194.
- Dixon, S.J.; Stockwell, B.R. The role of iron and reactive oxygen species in cell death. Nat. Chem. Biol. 2014, 10, 9–17.
- Latunde-Dada, G.O. Ferroptosis: Role of lipid peroxidation, iron and ferritinophagy. Biochim. Et Biophys. Acta Gen. Subj. 2017, 1861, 1893–1900.
- Iannielli, A.; Bido, S.; Folladori, L.; Segnali, A.; Cancellieri, C.; Maresca, A.; Massimino, L.; Rubio, A.; Morabito, G.; Caporali, L.; et al. Pharmacological Inhibition of Necroptosis Protects from Dopaminergic Neuronal Cell Death in Parkinson’s Disease Models. Cell Rep. 2018, 22, 2066–2079.
- Gonzalez, G.; Grúz, J.; D’Acunto, C.W.; Kaňovský, P.; Strnad, M. Cytokinin Plant Hormones Have Neuroprotective Activity in In Vitro Models of Parkinson’s Disease. Molecules 2021, 26, 361.
- Ashraf, A.; Clark, M.; So, P.-W. The Aging of Iron Man. Front. Aging Neurosci. 2018, 10, 65.
- McAllum, E.J.; Hare, D.J.; Volitakis, I.; McLean, C.A.; Bush, A.I.; Finkelstein, D.; Roberts, B.R. Regional iron distribution and soluble ferroprotein profiles in the healthy human brain. Prog. Neurobiol. 2020, 186, 101744.
- Zhang, Z.; Wei, T.; Hou, J.; Li, G.; Yu, S.; Xin, W. Iron-induced oxidative damage and apoptosis in cerebellar granule cells: Attenuation by tetramethylpyrazine and ferulic acid. Eur. J. Pharm. 2003, 467, 41–47.
- Hare, D.; Aror, M.; Jenkins, N.; Finkelstein, D.; Doble, P.; Bush, A. Is early-life iron exposure critical in neurodegeneration? Nat. Rev. Neurol. 2015, 11, 536–544.
- Ayton, S.; Lei, P.; Hare, D.; Duce, J.; George, J.; Adlard, P.; McLean, C.; Rogers, J.; Cherny, R.; Finkelstein, D.; et al. Parkinson’s disease iron deposition caused by nitric oxide-induced loss of β-amyloid precursor protein. J. Neurosci. 2015, 35, 3591–3597.
- Ayton, S.; Lei, P. Nigral iron elevation is an invariable feature of Parkinson’s disease and is a sufficient cause of neurodegeneration. Biomed Res. Int. 2014, 2014, 581256.
- Jenner, P.; Dexter, D.T.; Sian, J.; Schapira, A.H.; Marsden, C.D. Oxidative stress as a cause of nigral cell death in Parkinson’s disease and incidental Lewy body disease. The Royal Kings and Queens Parkinson’s Disease Research Group. Ann. Neurol. 1992, 32, S82-7.
- Costello, D.J.; Walsh, S.L.; Harrington, H.J.; Walsh, C.H. Concurrent hereditary haemochromatosis and idiopathic Parkinson’s disease: A case report series. J. Neurol. Neurosurg. Psychiatry 2004, 75, 631–633.
- Miyajima, H.; Takahashi, Y.; Kono, S. Aceruloplasminemia, an inherited disorder of iron metabolism. Biometals 2003, 16, 205–213.
- Nielsen, J.E.; Jensen, L.N.; Krabbe, K. Hereditary haemochromatosis: A case of iron accumulation in the basal ganglia associated with a parkinsonian syndrome. J. Neurol. Neurosurg. Psychiatry 1995, 59, 318–321.
- Borie, C.; Gasparini, F.; Verpillat, P.; Bonnet, A.-M.; Agid, Y.; Hetet, G.; Brice, A.; Dürr, A.; Grandchamp, B. French Parkinson’s disease genetic study group. Association study between iron-related genes polymorphisms and Parkinson’s disease. J. Neurol. 2002, 249, 801–804.
- Rhodes, S.; Buchanan, D.; Ahmed, I.; Taylor, K.; Loriot, M.-A.; Sinsheimer, J.; Bronstein, J.; Elbaz, A.; Mellick, G.; Rotter, J.; et al. Pooled analysis of iron-related genes in Parkinson’s disease: Association with transferrin. Neurobiol. Dis. 2014, 62, 172–178.
- Donovan, A.; Lima, C.A.; Pinkus, J.L.; Pinkus, G.S.; Zon, L.I.; Robine, S.; Andrews, N.C. The iron exporter ferroportin/Slc40a1 is essential for iron homeostasis. Cell Metab. 2005, 1, 191–200.
- Duce, J.A.; Tsatsanis, A.; Cater, M.A.; James, S.A.; Robb, E.; Wikhe, K.; Leong, S.L.; Perez, K.; Johanssen, T.; Greenough, M.A.; et al. Iron-export ferroxidase activity of β-amyloid precursor protein is inhibited by zinc in Alzheimer’s disease. Cell 2010, 142, 857–867.
- Lei, P.; Ayton, S.; I Finkelstein, D.; Spoerri, L.; Ciccotosto, G.D.; Wright, D.K.; Wong, B.X.W.; A Adlard, P.; A Cherny, R.; Lam, L.Q.; et al. Tau deficiency induces parkinsonism with dementia by impairing APP-mediated iron export. Nat. Med. 2012, 18, 291–295.
- Schulte, E.C.; Fukumori, A.; Mollenhauer, B.; Hor, H.; Arzberger, T.; Perneczky, R.; Kurz, A.; Diehl-Schmid, J.; Hüll, M.; Lichtner, P.; et al. Rare variants in β-Amyloid precursor protein (APP) and Parkinson’s disease. Eur. J. Hum. Genet. 2015, 23, 1328–1333.
- Edwards-Lee, T.; Ringman, J.M.; Chung, J.; Werner, J.; Morgan, A.; Hyslop, P.S.G.; Thompson, P.; Dutton, R.; Mlikotic, A.; Rogaeva, E.; et al. An African American family with early-onset Alzheimer disease and an APP (T714I) mutation. Neurology 2005, 64, 377–379.
- Halliday, G.; Brooks, W.; Arthur, H.; Creasey, H.; Broe, G. Further evidence for an association between a mutation in the APP gene and Lewy body formation. Neurosci. Lett. 1997, 227, 49–52.
- Rosenberg, C.K.; Pericak-Vance, M.A.; Saunders, A.M.; Gilbert, J.R.; Gaskell, P.C.; Hulette, C.M. Lewy body and Alzheimer pathology in a family with the amyloid-beta precursor protein APP717 gene mutation. Acta Neuropathol. 2000, 100, 145–152.
- Ayton, S.; Lei, P.; Duce, J.A.; Wong, B.X.; Sedjahtera, A.; Adlard, P.A.; Bush, A.I.; Finkelstein, D.I. Ceruloplasmin dysfunction and therapeutic potential for Parkinson’s disease. Ann. Neurol. 2013, 73, 554–559.
- Osaki, S.; Johnson, D.A.; Frieden, E. The possible significance of the ferrous oxidase activity of ceruloplasmin in normal human serum. J. Biol. Chem. 1966, 241, 2746–2751.
- Hochstrasser, H.; Bauer, P.; Walter, U.; Behnke, S.; Spiegel, J.; Csoti, I.; Zeiler, B.; Bornemann, A.; Pahnke, J.; Becker, G.; et al. Ceruloplasmin gene variations and substantia nigra hyperechogenicity in Parkinson’s disease. Neurology 2004, 63, 1912–1917.
- Kohno, S.; Miyajima, H.; Takahashi, Y.; Inoue, Y. Aceruloplasminemia with a novel mutation associated with parkinsonism. Neurogenetics 2000, 2, 237–238.
- Nagayama, H.; Hamamoto, M.; Ueda, M.; Nito, C.; Yamaguchi, H.; Katayama, Y. The effect of ascorbic acid on the pharmacokinetics of levodopa in elderly patients with Parkinson disease. Clin. Neuropharmacol. 2004, 27, 270–273.
- Chen, Q.; Espey, M.G.; Sun, A.Y.; Lee, J.-H.; Krishna, M.C.; Shacter, E.; Choyke, P.L.; Pooput, C.; Kirk, K.L.; Buettner, G.R.; et al. Ascorbate in pharmacologic concentrations selectively generates ascorbate radical and hydrogen peroxide in extracellular fluid in vivo. Proc. Natl. Acad. Sci. USA 2007, 104, 8749–8754.
- Sun, Y.; Pham, A.N.; Waite, T.D. The effect of vitamin C and iron on dopamine-mediated free radical generation: Implications to Parkinson’s disease. Dalton Trans 2018, 47, 4059–4069.
- Focarelli, F.; Giachino, A.; Waldron, K.J. Copper microenvironments in the human body define patterns of copper adaptation in pathogenic bacteria. PLoS Pathog. 2022, 18, e1010617.
- Tsang, T.; Davis, C.I.; Brady, D.C. Copper biology. Curr. Biol. 2021, 31, R421–R427.
- Tapiero, H.; Townsend, D.M.; Tew, K.D. Trace elements in human physiology and pathology. Copper. Biomed Pharm. 2003, 57, 386–398.
- Lutsenko, S.; Bhattacharjee, A.; Hubbard, A.L. Copper handling machinery of the brain. Metallomics 2010, 2, 596–608.
- Tarnacka, B.; Jopowicz, A.; Maślińska, M. Copper, Iron, and Manganese Toxicity in Neuropsychiatric Conditions. Int. J. Mol. Sci. 2021, 22, 7820.
- Amorós, R.; Murcia, M.; González, L.; Soler-Blasco, R.; Rebagliato, M.; Iñiguez, C.; Carrasco, P.; Vioque, J.; Broberg, K.; Levi, M.; et al. Maternal copper status and neuropsychological development in infants and preschool children. Int. J. Hyg. Environ. Health 2019, 222, 503–512.
- Gaier, E.D.; Eipper, B.A.; Mains, R.E. Copper signaling in the mammalian nervous system: Synaptic effects. J. Neurosci. Res. 2013, 91, 2–19.
- Gaetke, L.M.; Chow-Johnson, H.S.; Chow, C.K. Copper: Toxicological relevance and mechanisms. Arch. Toxicol. 2014, 88, 1929–1938.
- Da, S.; Rashed, L. Oxidative stress and genotoxicity among workers exposed to copper in a factory for non-ferrous industry in Egypt. Egypt. J. Occup. Med. 2019, 43, 1–15.
- Santos, S.; Silva, A.M.; Matos, M.; Monteiro, S.M.; Álvaro, A.R. Copper induced apoptosis in Caco-2 and Hep-G2 cells: Expression of caspases 3, 8 and 9, AIF and p53. Comp. Biochem. Physiol. Part C Toxicol. Pharm. 2016, 185–186, 138–146.
- Macip, S.; Igarashi, M.; Berggren, P.; Yu, J.; Lee, S.W.; Aaronson, S.A. Influence of induced reactive oxygen species in p53-mediated cell fate decisions. Mol. Cell. Biol. 2003, 23, 8576–8585.
- Narayanan, V.S.; Fitch, C.A.; Levenson, C.W. Tumor suppressor protein p53 mRNA and subcellular localization are altered by changes in cellular copper in human Hep G2 cells. J. Nutr. 2001, 131, 1427–1432.
- Gromadzka, G.; Tarnacka, B.; Flaga, A.; Adamczyk, A. Copper Dyshomeostasis in Neurodegenerative Diseases-Therapeutic Implications. Int. J. Mol. Sci. 2020, 21, 9259.
- Hozumi, I.; Hasegawa, T.; Honda, A.; Ozawa, K.; Hayashi, Y.; Hashimoto, K.; Yamada, M.; Koumura, A.; Sakurai, T.; Kimura, A.; et al. Patterns of levels of biological metals in CSF differ among neurodegenerative diseases. J. Neurol. Sci. 2011, 303, 95–99.
- Pall, H.S.; Williams, A.C.; Blake, D.R.; Lunec, J.; Gutteridge, J.M.; Hall, M.; Taylor, A. Raised cerebrospinal-fluid copper concentration in Parkinson’s disease. Lancet 1987, 2, 238–241.
- Genoud, S.; Senior, A.M.; Hare, D.J.; Double, K.L. Meta-Analysis of Copper and Iron in Parkinson’s Disease Brain and Biofluids. Mov. Disord. 2020, 35, 662–671.
- Santner, A.; Uversky, V.N. Metalloproteomics and metal toxicology of α-synuclein. Metallomics 2010, 2, 378–392.
- Gorell, J.M.; Johnson, C.C.; Rybicki, B.A.; Peterson, E.L.; Kortsha, G.X.; Brown, G.G.; Richardson, R.J. Occupational exposure to manganese, copper, lead, iron, mercury and zinc and the risk of Parkinson’s disease. Neurotoxicology 1999, 20, 239–247.
- Rybicki, B.A.; Johnson, C.C.; Uman, J.; Gorell, J.M. Parkinson’s disease mortality and the industrial use of heavy metals in Michigan. Mov. Disord. 1993, 8, 87–92.
- VanLandingham, J.W.; Tassabehji, N.M.; Somers, R.C.; Levenson, C.W. Expression profiling of p53-target genes in copper-mediated neuronal apoptosis. Neuromolecular Med. 2005, 7, 311–324.
- Lu, Y.; Prudent, M.; Fauvet, B.; Lashuel, H.A.; Girault, H.H. Phosphorylation of α-Synuclein at Y125 and S129 alters its metal binding properties: Implications for understanding the role of α-Synuclein in the pathogenesis of Parkinson’s Disease and related disorders. ACS Chem. Neurosci. 2011, 2, 667–675.
- Miotto, M.C.; Rodriguez, E.E.; Valiente-Gabioud, A.A.; Torres-Monserrat, V.; Binolfi, A.; Quintanar, L.; Zweckstetter, M.; Griesinger, C.; Fernández, C.O. Site-specific copper-catalyzed oxidation of α-synuclein: Tightening the link between metal binding and protein oxidative damage in Parkinson’s disease. Inorg. Chem. 2014, 53, 4350–4358.
- Anandhan, A.; Rodriguez-Rocha, H.; Bohovych, I.; Griggs, A.M.; Zavala-Flores, L.; Reyes-Reyes, E.M.; Seravalli, J.; Stanciu, L.A.; Lee, J.; Rochet, J.-C.; et al. Overexpression of alpha-synuclein at non-toxic levels increases dopaminergic cell death induced by copper exposure via modulation of protein degradation pathways. Neurobiol. Dis. 2015, 81, 76–92.
- Letelier, M.E.; FaúnNdez, M.; Jara-Sandoval, J.; Molina-Berrã os, A.; Cortãés-Troncoso, J.; Aracena-Parks, P.; Marín-Catalán, R. Mechanisms underlying the inhibition of the cytochrome P450 system by copper ions. J. Appl. Toxicol. 2009, 29, 695–702.
- Letelier, M.E.; Lepe, A.M.; Faúndez, M.; Salazar, J.; Marín, R.; Aracena, P.; Speisky, H. Possible mechanisms underlying copper-induced damage in biological membranes leading to cellular toxicity. Chem. Biol. Interact 2005, 151, 71–82.
- Kim, M.J.; Oh, S.B.; Kim, J.; Kim, K.; Ryu, H.S.; Kim, M.S.; Ayton, S.; Bush, A.I.; Lee, J.Y.; Chung, S.J. Association of metals with the risk and clinical characteristics of Parkinson’s disease. Park. Relat Disord 2018, 55, 117–121.
- Doguer, C.; Ha, J.H.; Collins, J.F. Intersection of Iron and Copper Metabolism in the Mammalian Intestine and Liver. Compr. Physiol. 2018, 8, 1433–1461.

