Biometals are all metal ions that are essential for all living organisms. About 40% of all enzymes with known structures require biometals to function correctly. The main target of damage by biometals is the central nervous system (CNS). Biometal dysregulation (metal deficiency or overload) is related to pathological processes. Chronic occupational and environmental exposure to biometals, including iron and copper, is related to an increased risk of developing Parkinson’s disease (PD). Indeed, biometals have been shown to induce a dopaminergic neuronal loss in the substantia nigra. Although the etiology of PD is still unknown, oxidative stress dysregulation, mitochondrial dysfunction, and inhibition of both the ubiquitin–proteasome system (UPS) and autophagy are related to dopaminergic neuronal death.
- redox-active
- biometals
- iron
- copper
- Parkinson’s disease
1. Metals, Parkinson’s Disease, and Oxidative Stress
2. Redox-Active Metals’ Role in Dopaminergic Neuronal Death
2.1. Iron
Iron is the most abundant metal on Earth, and almost all organisms have evolved to use this ubiquitous transition metal [25]. This metal is essential for the human body’s proper functioning. Iron is vital for oxygen transport (bound to hemoglobin), oxidative phosphorylation (bound to cytochrome C), neurotransmitter synthesis, myelin formation, and regulation of the biosynthesis of proteins such as ferritin and transferrin receptor (to store or mobilize iron) through iron-response proteins binding to iron-responsive elements at mRNA level [26][27][28]. In contrast, unbound iron causes cell toxicity as it can trigger a series of highly oxidative and toxic reactions [29]; this occurs when the iron concentrations exceed the binding capacity of transferrin [30]. Iron has a wide range of oxidative states, Fe2+ (ferrous) and Fe3+ (ferric) being the most common in biological environments [31]. Fe2+ binding to proteins is very unstable [32]. Iron’s neurotoxic effect has been related to the divalent metal ion transporter 1 (DMT1) overexpression, which imports iron into the cell, and can also be inhibited by H-ferritin [33]. Moreover, S-nitrosylation (SNO) of DMT1 cysteine thiol enhances Mn2+ and Fe2+ uptake [34]. Additionally, SNO-DMT1 has been detected in the post-mortem substantia nigra of PD patients [34]. The redox state of iron determines its role in cytotoxic reactions [35]. In a healthy brain, iron is distributed in a specific pattern by region and cell type. It is abundant in the substantia nigra and the basal ganglia, which are rich in dopaminergic neurons [36]. For over four decades, changes in iron and ferritin levels have been described in the brain of PD patients [15]. Several mechanisms are associated with iron-induced dopaminergic cell death, including (1) Fenton redox-reactions producing hydroxyl radicals [37]; (2) DA oxidative deamination, which is catalyzed by monoamine oxidase B (MAO-B) and regulated by Fe2+ and Fe3+ [38][39]; (3) 6-hydroxydopamine neurotoxic formation through DA metabolites reaction with iron and H2O2 [40]; and (4) increased rate of iron-induced α-synuclein fibril formation [21]. Iron takes part in the Fenton reaction producing free radicals; Fe2+ reacts with H2O2 or lipid peroxides to generate Fe3+, hydroxyl ion (OH–), and OH• or lipid radicals, which may lead to oxidative damage of macromolecules [27][41]. The Haber–Weiss reaction is where hydroxyl ion and hydroxyl radical are generated from the reaction of H2O2 and O2•– catalyzed by iron [42]. Additionally, the formation and accumulation of OH• lead to activation of the mitochondrial permeability transition pore (mPTP), which temporarily opens and increases ROS, provoking long-lasting activation and cell death. The latter is triggered by a decrease of ATP production, mitochondrial swelling, and rupture of the outer mitochondrial membrane, with subsequent release of mitochondrial death factors such as cytochrome C to the cytosolic compartment activating cell death by apoptosis [43]. Moreover, p53 is involved in mitochondrial dysfunction and oxidative stress mediated by Fe2+ in neuronal synaptic terminals [44]. Interestingly, lysosomes contain a redox-active iron pool derived from iron-rich macromolecules and cellular organelles, such as ferritin and mitochondria [45][46]. Most iron is found in a non-redox active form bound to ferritin. Ferritin degradation inside lysosomes during autophagy may be an intracellular redox-active iron source [47]. Next, H2O2 diffuses into lysosomes and reacts with the iron species through the Fenton and Fenton-like reactions, resulting in hydroxyl radical generation [45]. Intriguingly, defective mitochondria and lysosomes may promote receptor-interacting protein kinase 1 (RIPK1) activation, making cells susceptible to necroptosis [48]. Recently, necroptosis’s partial contribution to iron-mediated toxicity was demonstrated by using iron chelator deferoxamine (DFO) and the necroptosis inhibitor necrostatin 1 (NEC-1), significantly reducing cell death rates in the glutamate-induced model in vitro [49]. Iron accumulation in the brain is age-dependent, having the lowest levels at birth and a marked increase with age [50]. Brain regions associated with motor function have a high concentration of iron [50], and its levels in the whole brain are around 35.6–54.2 µg/g [51]. Its accumulation in the brain affects neurons. Exposure of neurons to iron induces oxidative stress, causing lipid peroxidation and DNA damage, which leads to caspase-dependent apoptotic cell death [52]. Several key mediators of ferroptosis have previously been implicated in PD pathogenesis. The SNpc is an iron-rich, dopamine (DA)-producing midbrain nucleus, which probably explains why it has a high risk of suffering neuronal death [53][54], mainly when iron accumulates, representing a PD feature [15][55]. Iron produces hydroxyl radicals with subsequent dopamine oxidation, likely contributing to an oxidative environment that increases the loss of nigral dopaminergic neurons in PD patients [56]. Moreover, genetic disorders that result in brain iron dyshomeostasis often cause Parkinsonism [57][58][59], demonstrating increased iron’s potential to contribute to PD pathogenesis. Indeed, mutations in several proteins involved in iron transport, increasing iron uptake and decreasing its export, are linked to PD. Mutant forms of transferrin, a critical protein for neuronal iron uptake, are associated with increased susceptibility to PD [60][61]. These data suggest that the iron uptake mechanism is overactive in these patients resulting in increased neuronal iron accumulation. Conversely, mutations in transferrin receptor 2 (TfR2) [61] are associated with a protective effect in PD, potentially due to reduced iron uptake. Neuronal iron export occurs via a transmembrane ion channel, ferroportin [62], and the Alzheimer’s disease (AD)-implicated amyloid precursor protein (APP) stabilizes ferroportin expression on the membrane to promote iron efflux [63]. In contrast, loss of APP membrane function results in impaired iron efflux and consequent neuronal iron retention [64]. Indeed, several rare variants of APP predispose individuals to PD, and several studies of familial AD indicate APP mutations are associated with Parkinsonism and LB formation [65][66][67][68]. Deficits in iron export in PD were further identified in the substantia nigra, with a significant depletion in APP expression levels independently of cell loss and an 80% decrease in ceruloplasmin (CP) activity [54][69]. Ceruloplasmin also has a ferrous oxidase activity and enables iron export by converting Fe2+ to Fe3+ [70], which is then bound to and removed by transferrin. Several point mutations in the CP-encoding gene are significantly associated with PD [71] and Parkinsonism [58][72], indicating that CP-mediated iron homeostasis is also likely involved in PD pathogenesis. Vitamin C, or ascorbic acid, has been shown to improve the absorption of Levodopa in some PD patients with poor Levodopa bioavailability [73]. However, it might be toxic as it loses one electron and forms an ascorbate radical (Asc•–). The electron can reduce metal ions such as iron and copper. The acidic extracellular environment favors the reduction of protein-centered metal, represented as Fe3+ reduction to Fe2+. Subsequently, Fe2+ donates an electron to O2 forming O2•– with subsequent dismutation to H2O2 [74]. Moreover, vitamin C administration may aggravate PD progression due to the possible peroxidation of Fe2+ bound to Asc•–. Therefore, combined vitamin C therapy for ROS scavenging and an iron chelator to sequester the metal may be a promising PD treatment option, reducing the toxicity induced by DA-derived quinones [75].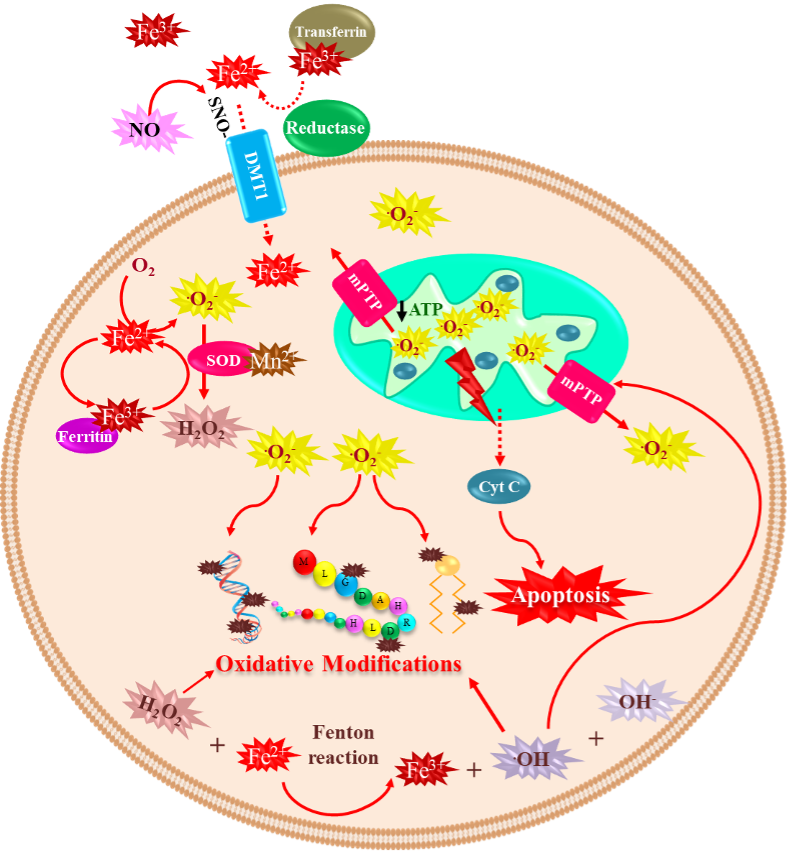
2.2. Copper
Copper is a trace element that constitutes 70 parts per million of the Earth’s crust. However, it is an essential micronutrient found in small amounts in tissues and cells, with a high concentration in the kidney, liver, and brain [76]. This metal functions as an essential cofactor and is required for structural and catalytic proprieties of more than 30 necessary enzymes; among them are ceruloplasmin, cytochrome oxidase, lysine oxidase, dopamine-hydroxylase, ascorbate oxidase, tyrosinase, and Cu/Zn SOD [77]. In living organisms, copper is mainly found oxidized (Cu2+) and reduced (Cu+) [78]. After the liver, the brain is the organ that accumulates the most significant amount of copper, reaching contents between 2.9 to 10 µg/g wet weight [79], and it is distributed differently in each region. The regions with the highest concentrations of copper are the substantia nigra, cerebellum, hippocampus, and hypothalamus [80]. Copper plays a crucial role in essential processes in CNS, such as brain development [81], antioxidant defense, synaptic transmission [82], and acting as an enzyme cofactor with oxidoreductase activity [77]. Notwithstanding, like any redox-active metal, copper becomes toxic when its intracellular accumulation is excessive, facilitating the formation of ROS and apoptotic processes [83]. When the cell is exposed to oxidative stress or copper, DNA damage and p53 expression are induced [84][85]. Furthermore, p53 undergoes oligomerization and phosphorylation to be translocated into the nucleus to induce genes such as BAX (BCL2 Associated X) and PUMA (p53 upregulated modulator of apoptosis) and subsequent release of cytochrome C into the cytosol to initiate apoptosis [86][87]. Therefore, copper plays a vital role in many diseases, such as Menkes disease, where copper is abnormally low in the brain. Conversely, in Wilson’s disease, the damage is caused by an excess of copper stored in brain tissue. Moreover, some neurodegenerative disorders such as AD, amyotrophic lateral sclerosis (ALS), prion disease, and PD have been linked to copper dyshomeostasis [88]. Increased copper levels have been reported in the cerebrospinal fluid and blood of PD patients [89][90]. However, a recent meta-analysis reported decreased copper levels in the substantia nigra of PD patients compared to healthy age-matched subjects [91]. Nevertheless, it has been demonstrated that chronic occupational exposure to copper increases the risk of developing PD [92][93][94]. Copper toxicity affects the basal ganglia and frontal cortex inducing Parkinson-like symptoms and cognitive deficits. The mechanism of copper-mediated toxicity includes cell cycle arrest via the upregulation of p21 (Cyclin Dependent Kinase Inhibitor 1A), reprimo (involved in regulating p53-dependent G2 arrest of the cell cycle and coded by RPRM gene), stathmin (microtubule destabilizing protein coded by STMN1 gene), and Tp53INP1 (Tumor Protein P53 Inducible Nuclear Protein 1). Additionally, stat-3 (Signal Transducer and Activator of Transcription 3), hsp70 (Heat Shock Protein 70), and hsp27 (Heat Shock Protein 27) are increased in an attempt to survive. Finally, p53-dependent and independent apoptosis are triggered, where IGFBP-6 (insulin-like growth factor binding protein-6), glutathione peroxidase, BCL-2, RB-1, PUMA, and several members of the redox-active PIG family of proteins, play a role [95]. Moreover, copper binding to α-synuclein increases oxidative stress and α-synuclein phosphorylation and accelerates the protein aggregation process [22][96][97]. In the neuroblastoma cell line SK-N-SH with dopaminergic phenotype, copper transporter protein 1 (Ctr1) overexpression led to intracellular glutathione depletion and potentiated the caspase-3-dependent-cell death induced by copper, indicating that copper’s toxicity is due to alterations in its intracellular homeostasis. In addition, copper-induced oxidative stress was primarily localized in the cytosol, and Nrf2 was upregulated to mediate an antioxidant response. In addition, copper increased protein ubiquitination, AMPK-Ulk1 signaling, p62, and Atg5-dependent autophagy as a protective mechanism [98]. The release of redox-active copper ions from copper-binding proteins and its binding to thiol or amine groups of cysteinyl and histidinyl residues of globular proteins, including enzymes, may result in conformational changes leading to its inactivation [99][100]. Paradoxically, some clinical trials report decreased circulating copper levels in PD patients compared to healthy controls [101], which may occur, because by binding to ceruloplasmin, copper stimulates ferroxidase activity and participates in iron homeostasis. Therefore, low levels of copper can indirectly generate toxicity by altering iron concentrations [102].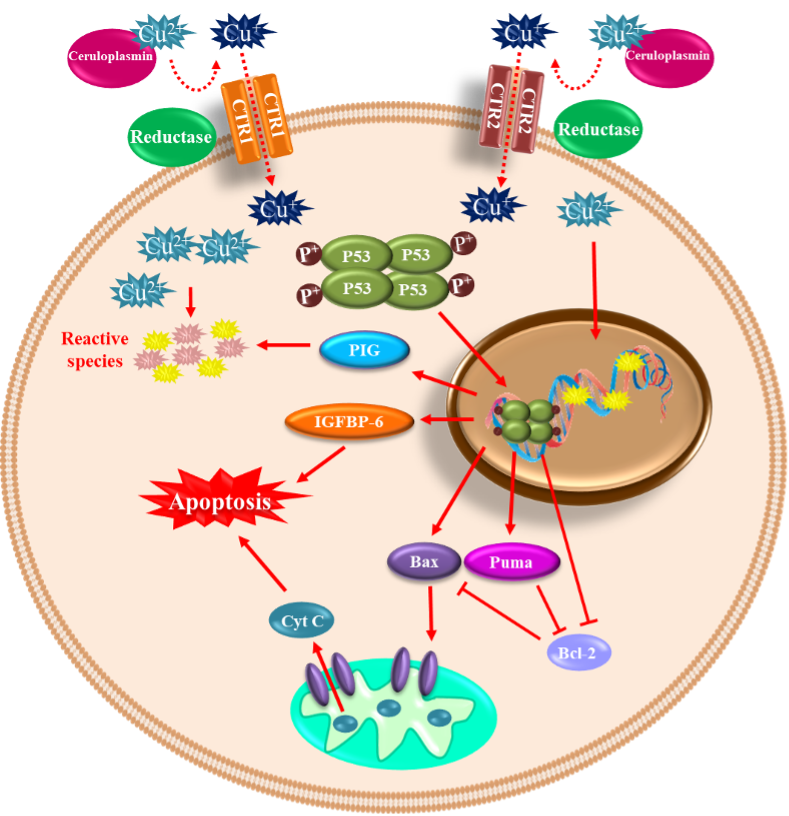 Figure 2. Copper-mediated neuronal cell death. Copper can be found in its oxidized (Cu2+) and reduced (Cu+) forms within the cells. The oxidized form of copper is bound to proteins, and when reduced by a reductase enzyme, it enters the cell through the transporters CTR1 and CTR2. Once inside and in high concentration, it induces DNA damage and p53 expression. p53 undergoes oligomerization/phosphorylation and is translocated into the nucleus to induce BAX and PUMA, with the consequent release of cytochrome C into the cytosol to initiate apoptosis. p53-dependent and independent apoptosis is triggered, where insulin-like growth factor binding protein-6 (IGFBP-6) and PIG proteins (prooxidant proteins), among other players, are involved.
In summary, nearly 40% of ourthe proteins need a biometal as a cofactor to fulfill their function. Therefore, it is crucial to understand the transition metals’ role in health and disease because their dyshomeostasis (deficiency or overloading) is closely related to different disorders and mainly to neurodegeneration. Research in this field has found that biometals are tightly regulated because a narrow unbalance provokes diseases such as PD. Therefore, understanding the complexity of the interaction between transition metals and proteins can shed light on possible neurodegeneration biomarkers for preventing neuronal cell death. Hopefully, neurodegeneration may be achieved by either supplementing transition metals when there is a deficiency or using chelating agents to avoid metal overload that induces neuronal cell death in PD.
Figure 2. Copper-mediated neuronal cell death. Copper can be found in its oxidized (Cu2+) and reduced (Cu+) forms within the cells. The oxidized form of copper is bound to proteins, and when reduced by a reductase enzyme, it enters the cell through the transporters CTR1 and CTR2. Once inside and in high concentration, it induces DNA damage and p53 expression. p53 undergoes oligomerization/phosphorylation and is translocated into the nucleus to induce BAX and PUMA, with the consequent release of cytochrome C into the cytosol to initiate apoptosis. p53-dependent and independent apoptosis is triggered, where insulin-like growth factor binding protein-6 (IGFBP-6) and PIG proteins (prooxidant proteins), among other players, are involved.
In summary, nearly 40% of ourthe proteins need a biometal as a cofactor to fulfill their function. Therefore, it is crucial to understand the transition metals’ role in health and disease because their dyshomeostasis (deficiency or overloading) is closely related to different disorders and mainly to neurodegeneration. Research in this field has found that biometals are tightly regulated because a narrow unbalance provokes diseases such as PD. Therefore, understanding the complexity of the interaction between transition metals and proteins can shed light on possible neurodegeneration biomarkers for preventing neuronal cell death. Hopefully, neurodegeneration may be achieved by either supplementing transition metals when there is a deficiency or using chelating agents to avoid metal overload that induces neuronal cell death in PD.
References
- McCord, J.M. The evolution of free radicals and oxidative stress. Am. J. Med. 2000, 108, 652–659.
- Martemucci, G.; Costagliola, C.; Mariano, M.; D’Andrea, L.; Napolitano, P.; D’Alessandro, A.G. Free Radical Properties, Source and Targets, Antioxidant Consumption and Health. Oxygen 2022, 2, 48–78.
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative Stress: Harms and Benefits for Human Health. Oxidative Med. Cell. Longev. 2017, 2017, 8416763.
- Gilgun-Sherki, Y.; Melamed, E.; Offen, D. Oxidative stress induced-neurodegenerative diseases: The need for antioxidants that penetrate the blood brain barrier. Neuropharmacology 2001, 40, 959–975.
- Navarro-Yepes, J.; Burns, M.; Anandhan, A.; Khalimonchuk, O.; del Razo, L.M.; Quintanilla-Vega, B.; Pappa, A.; Panayiotidis, M.; Franco, R. Oxidative stress, redox signaling, and autophagy: Cell death versus survival. Antioxid. Redox Signal. 2014, 21, 66–85.
- Kurutas, E.B. The importance of antioxidants which play the role in cellular response against oxidative/nitrosative stress: Current state. Nutr. J. 2016, 15, 71.
- Forman, H.J.; Zhang, H. Targeting oxidative stress in disease: Promise and limitations of antioxidant therapy. Nat. Rev. Drug Discov. 2021, 20, 689–709.
- Stykel, M.G.; Ryan, S.D. Nitrosative stress in Parkinson’s disease. NPJ Parkinsons Dis. 2022, 8, 104.
- Hartmann, A. Postmortem studies in Parkinson’s disease. Dialogues Clin. Neurosci. 2004, 6, 281–293.
- Farzam, A.; Chohan, K.; Strmiskova, M.; Hewitt, S.J.; Park, D.S.; Pezacki, J.P.; Özcelik, D. A functionalized hydroxydopamine quinone links thiol modification to neuronal cell death. Redox Biol. 2020, 28, 101377.
- Cobley, J.N.; Fiorello, M.L.; Bailey, D.M. 13 reasons why the brain is susceptible to oxidative stress. Redox Biol. 2018, 15, 490–503.
- Kish, S.J.; Morito, C.; Hornykiewicz, O. Glutathione peroxidase activity in Parkinson’s disease brain. Neurosci. Lett. 1985, 58, 343–346.
- Chen, C.; Turnbull, D.M.; Reeve, A.K. Mitochondrial Dysfunction in Parkinson’s Disease-Cause or Consequence? Biology 2019, 8, 38.
- Ball, N.; Teo, W.-P.; Chandra, S.; Chapman, J. Parkinson’s Disease and the Environment. Front. Neurol. 2019, 10, 218.
- Dexter, D.T.; Wells, F.R.; Lees, A.J.; Agid, F.; Agid, Y.; Jenner, P.; Marsden, C.D. Increased nigral iron content and alterations in other metal ions occurring in brain in Parkinson’s disease. J. Neurochem. 1989, 52, 1830–1836.
- Nakamura, K.; Bindokas, V.P.; Marks, J.D.; Wright, D.A.; Frim, D.M.; Miller, R.J.; Kang, U.J. The selective toxicity of 1-methyl-4-phenylpyridinium to dopaminergic neurons: The role of mitochondrial complex I and reactive oxygen species revisited. Mol. Pharm. 2000, 58, 271–278.
- Obata, T. Dopamine efflux by MPTP and hydroxyl radical generation. J. Neural Transm. 2002, 109, 1159–1180.
- Hoffman, C.; Aballay, A. Role of neurons in the control of immune defense. Curr. Opin. Immunol. 2019, 60, 30–36.
- Lesage, S.; Brice, A. Parkinson’s disease: From monogenic forms to genetic susceptibility factors. Hum. Mol. Genet. 2009, 18, R48–R59.
- Shults, C.W. Lewy bodies. Proc. Natl. Acad. Sci. USA 2006, 103, 1661–1668.
- Uversky, V.N.; Li, J.; Fink, A.L. Metal-triggered structural transformations, aggregation, and fibrillation of human alpha-synuclein. A possible molecular NK between Parkinson’s disease and heavy metal exposure. J. Biol. Chem. 2001, 276, 44284–44296.
- Liu, L.L.; Franz, K.J. Phosphorylation-dependent metal binding by alpha-synuclein peptide fragments. J. Biol. Inorg. Chem. 2007, 12, 234–247.
- Liu, L.L.; Franz, K.J. Phosphorylation of an alpha-synuclein peptide fragment enhances metal binding. J. Am. Chem. Soc. 2005, 127, 9662–9663.
- Garcia-Garcia, A.; Rodriguez-Rocha, H.; Madayiputhiya, N.; Pappa, A.; Panayiotidis, M.I.; Franco, R. Biomarkers of protein oxidation in human disease. Curr. Mol. Med. 2012, 12, 681–697.
- Dlouhy, A.C.; Outten, C.E. The iron metallome in eukaryotic organisms. Met. Ions Life Sci. 2013, 12, 241–278.
- Casey, J.L.; Hentze, M.W.; Koeller, D.M.; Caughman, S.W.; Rouault, T.A.; Klausner, R.D.; Harford, J.B. Iron-responsive elements: Regulatory RNA sequences that control mRNA levels and translation. Science 1988, 240, 924–928.
- Hentze, M.W.; Muckenthaler, M.U.; Andrews, N.C. Balancing acts: Molecular control of mammalian iron metabolism. Cell 2004, 117, 285–297.
- Arosio, P.; Elia, L.; Poli, M. Ferritin, cellular iron storage and regulation. IUBMB Life 2017, 69, 414–422.
- Bresgen, N.; Eckl, P.M. Oxidative stress and the homeodynamics of iron metabolism. Biomolecules 2015, 5, 808–847.
- Siah, C.W.; Ombiga, J.; Adams, L.A.; Trinder, D.; Olynyk, J.K. Normal iron metabolism and the pathophysiology of iron overload disorders. Clin. Biochem. Rev. 2006, 27, 5–16.
- Núñez, M.T.; Urrutia, P.; Mena, N.; Aguirre, P.; Tapia, V.; Salazar, J. Iron toxicity in neurodegeneration. Biometals 2012, 25, 761–776.
- Cotruvo, J.J.A.; Stubbe, J. Metallation and mismetallation of iron and manganese proteins in vitro and in vivo: The class I ribonucleotide reductases as a case study. Metallomics 2012, 4, 1020–1036.
- Zhu, W.; Li, X.; Xie, W.; Luo, F.; Kaur, D.; Andersen, J.K.; Jankovic, J.; Le, W. Genetic iron chelation protects against proteasome inhibition-induced dopamine neuron degeneration. Neurobiol. Dis. 2010, 37, 307–313.
- Liu, C.; Zhang, C.-W.; Lo, S.Q.; Ang, S.T.; Chew, K.C.M.; Yu, D.; Chai, B.H.; Tan, B.; Tsang, F.; Tai, Y.K.; et al. S-Nitrosylation of Divalent Metal Transporter 1 Enhances Iron Uptake to Mediate Loss of Dopaminergic Neurons and Motoric Deficit. J. Neurosci. 2018, 38, 8364–8377.
- Yarjanli, Z.; Ghaedi, K.; Esmaeili, A.; Rahgozar, S.; Zarrabi, A. Iron oxide nanoparticles may damage to the neural tissue through iron accumulation, oxidative stress, and protein aggregation. BMC Neurosci. 2017, 18, 51.
- Ramos, P.; Santos, A.; Pinto, N.R.; Mendes, R.; Magalhães, T.; Almeida, A. Iron levels in the human brain: A post-mortem study of anatomical region differences and age-related changes. J. Trace Elem. Med. Biol. 2014, 28, 13–17.
- Linert, W.; Jameson, G.N. Redox reactions of neurotransmitters possibly involved in the progression of Parkinson’s Disease. J. Inorg. Biochem. 2000, 79, 319–326.
- Youdim, M.B.; Riederer, P.F. A review of the mechanisms and role of monoamine oxidase inhibitors in Parkinson’s disease. Neurology 2004, 63 (Suppl. 2), S32-5.
- Lu, H.; Chen, J.; Huang, H.; Zhou, M.; Zhu, Q.; Yao, S.Q.; Chai, Z.; Hu, Y. Iron modulates the activity of monoamine oxidase B in SH-SY5Y cells. Biometals 2017, 30, 599–607.
- Napolitano, A.; Manini, P.; d’Ischia, M. Oxidation chemistry of catecholamines and neuronal degeneration: An update. Curr. Med. Chem. 2011, 18, 1832–1845.
- Barbusinski, K. Toxicity of Industrial Wastewater Treated by Fenton’s Reagent. Pol. J. Environ. Stud. 2005, 14, 11–16.
- Kehrer, J.P. The Haber-Weiss reaction and mechanisms of toxicity. Toxicology 2000, 149, 43–50.
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950.
- Gilman, C.P.; Chan, S.L.; Guo, Z.; Zhu, X.; Greig, N.; Mattson, M.P. p53 is present in synapses where it mediates mitochondrial dysfunction and synaptic degeneration in response to DNA damage, and oxidative and excitotoxic insults. Neuromolecular Med. 2003, 3, 159–172.
- Koskenkorva-Frank, T.S.; Weiss, G.; Koppenol, W.H.; Burckhardt, S. The complex interplay of iron metabolism, reactive oxygen species, and reactive nitrogen species: Insights into the potential of various iron therapies to induce oxidative and nitrosative stress. Free Radic. Biol. Med. 2013, 65, 1174–1194.
- Dixon, S.J.; Stockwell, B.R. The role of iron and reactive oxygen species in cell death. Nat. Chem. Biol. 2014, 10, 9–17.
- Latunde-Dada, G.O. Ferroptosis: Role of lipid peroxidation, iron and ferritinophagy. Biochim. Et Biophys. Acta Gen. Subj. 2017, 1861, 1893–1900.
- Iannielli, A.; Bido, S.; Folladori, L.; Segnali, A.; Cancellieri, C.; Maresca, A.; Massimino, L.; Rubio, A.; Morabito, G.; Caporali, L.; et al. Pharmacological Inhibition of Necroptosis Protects from Dopaminergic Neuronal Cell Death in Parkinson’s Disease Models. Cell Rep. 2018, 22, 2066–2079.
- Gonzalez, G.; Grúz, J.; D’Acunto, C.W.; Kaňovský, P.; Strnad, M. Cytokinin Plant Hormones Have Neuroprotective Activity in In Vitro Models of Parkinson’s Disease. Molecules 2021, 26, 361.
- Ashraf, A.; Clark, M.; So, P.-W. The Aging of Iron Man. Front. Aging Neurosci. 2018, 10, 65.
- McAllum, E.J.; Hare, D.J.; Volitakis, I.; McLean, C.A.; Bush, A.I.; Finkelstein, D.; Roberts, B.R. Regional iron distribution and soluble ferroprotein profiles in the healthy human brain. Prog. Neurobiol. 2020, 186, 101744.
- Zhang, Z.; Wei, T.; Hou, J.; Li, G.; Yu, S.; Xin, W. Iron-induced oxidative damage and apoptosis in cerebellar granule cells: Attenuation by tetramethylpyrazine and ferulic acid. Eur. J. Pharm. 2003, 467, 41–47.
- Hare, D.; Aror, M.; Jenkins, N.; Finkelstein, D.; Doble, P.; Bush, A. Is early-life iron exposure critical in neurodegeneration? Nat. Rev. Neurol. 2015, 11, 536–544.
- Ayton, S.; Lei, P.; Hare, D.; Duce, J.; George, J.; Adlard, P.; McLean, C.; Rogers, J.; Cherny, R.; Finkelstein, D.; et al. Parkinson’s disease iron deposition caused by nitric oxide-induced loss of β-amyloid precursor protein. J. Neurosci. 2015, 35, 3591–3597.
- Ayton, S.; Lei, P. Nigral iron elevation is an invariable feature of Parkinson’s disease and is a sufficient cause of neurodegeneration. Biomed Res. Int. 2014, 2014, 581256.
- Jenner, P.; Dexter, D.T.; Sian, J.; Schapira, A.H.; Marsden, C.D. Oxidative stress as a cause of nigral cell death in Parkinson’s disease and incidental Lewy body disease. The Royal Kings and Queens Parkinson’s Disease Research Group. Ann. Neurol. 1992, 32, S82-7.
- Costello, D.J.; Walsh, S.L.; Harrington, H.J.; Walsh, C.H. Concurrent hereditary haemochromatosis and idiopathic Parkinson’s disease: A case report series. J. Neurol. Neurosurg. Psychiatry 2004, 75, 631–633.
- Miyajima, H.; Takahashi, Y.; Kono, S. Aceruloplasminemia, an inherited disorder of iron metabolism. Biometals 2003, 16, 205–213.
- Nielsen, J.E.; Jensen, L.N.; Krabbe, K. Hereditary haemochromatosis: A case of iron accumulation in the basal ganglia associated with a parkinsonian syndrome. J. Neurol. Neurosurg. Psychiatry 1995, 59, 318–321.
- Borie, C.; Gasparini, F.; Verpillat, P.; Bonnet, A.-M.; Agid, Y.; Hetet, G.; Brice, A.; Dürr, A.; Grandchamp, B. French Parkinson’s disease genetic study group. Association study between iron-related genes polymorphisms and Parkinson’s disease. J. Neurol. 2002, 249, 801–804.
- Rhodes, S.; Buchanan, D.; Ahmed, I.; Taylor, K.; Loriot, M.-A.; Sinsheimer, J.; Bronstein, J.; Elbaz, A.; Mellick, G.; Rotter, J.; et al. Pooled analysis of iron-related genes in Parkinson’s disease: Association with transferrin. Neurobiol. Dis. 2014, 62, 172–178.
- Donovan, A.; Lima, C.A.; Pinkus, J.L.; Pinkus, G.S.; Zon, L.I.; Robine, S.; Andrews, N.C. The iron exporter ferroportin/Slc40a1 is essential for iron homeostasis. Cell Metab. 2005, 1, 191–200.
- Duce, J.A.; Tsatsanis, A.; Cater, M.A.; James, S.A.; Robb, E.; Wikhe, K.; Leong, S.L.; Perez, K.; Johanssen, T.; Greenough, M.A.; et al. Iron-export ferroxidase activity of β-amyloid precursor protein is inhibited by zinc in Alzheimer’s disease. Cell 2010, 142, 857–867.
- Lei, P.; Ayton, S.; I Finkelstein, D.; Spoerri, L.; Ciccotosto, G.D.; Wright, D.K.; Wong, B.X.W.; A Adlard, P.; A Cherny, R.; Lam, L.Q.; et al. Tau deficiency induces parkinsonism with dementia by impairing APP-mediated iron export. Nat. Med. 2012, 18, 291–295.
- Schulte, E.C.; Fukumori, A.; Mollenhauer, B.; Hor, H.; Arzberger, T.; Perneczky, R.; Kurz, A.; Diehl-Schmid, J.; Hüll, M.; Lichtner, P.; et al. Rare variants in β-Amyloid precursor protein (APP) and Parkinson’s disease. Eur. J. Hum. Genet. 2015, 23, 1328–1333.
- Edwards-Lee, T.; Ringman, J.M.; Chung, J.; Werner, J.; Morgan, A.; Hyslop, P.S.G.; Thompson, P.; Dutton, R.; Mlikotic, A.; Rogaeva, E.; et al. An African American family with early-onset Alzheimer disease and an APP (T714I) mutation. Neurology 2005, 64, 377–379.
- Halliday, G.; Brooks, W.; Arthur, H.; Creasey, H.; Broe, G. Further evidence for an association between a mutation in the APP gene and Lewy body formation. Neurosci. Lett. 1997, 227, 49–52.
- Rosenberg, C.K.; Pericak-Vance, M.A.; Saunders, A.M.; Gilbert, J.R.; Gaskell, P.C.; Hulette, C.M. Lewy body and Alzheimer pathology in a family with the amyloid-beta precursor protein APP717 gene mutation. Acta Neuropathol. 2000, 100, 145–152.
- Ayton, S.; Lei, P.; Duce, J.A.; Wong, B.X.; Sedjahtera, A.; Adlard, P.A.; Bush, A.I.; Finkelstein, D.I. Ceruloplasmin dysfunction and therapeutic potential for Parkinson’s disease. Ann. Neurol. 2013, 73, 554–559.
- Osaki, S.; Johnson, D.A.; Frieden, E. The possible significance of the ferrous oxidase activity of ceruloplasmin in normal human serum. J. Biol. Chem. 1966, 241, 2746–2751.
- Hochstrasser, H.; Bauer, P.; Walter, U.; Behnke, S.; Spiegel, J.; Csoti, I.; Zeiler, B.; Bornemann, A.; Pahnke, J.; Becker, G.; et al. Ceruloplasmin gene variations and substantia nigra hyperechogenicity in Parkinson’s disease. Neurology 2004, 63, 1912–1917.
- Kohno, S.; Miyajima, H.; Takahashi, Y.; Inoue, Y. Aceruloplasminemia with a novel mutation associated with parkinsonism. Neurogenetics 2000, 2, 237–238.
- Nagayama, H.; Hamamoto, M.; Ueda, M.; Nito, C.; Yamaguchi, H.; Katayama, Y. The effect of ascorbic acid on the pharmacokinetics of levodopa in elderly patients with Parkinson disease. Clin. Neuropharmacol. 2004, 27, 270–273.
- Chen, Q.; Espey, M.G.; Sun, A.Y.; Lee, J.-H.; Krishna, M.C.; Shacter, E.; Choyke, P.L.; Pooput, C.; Kirk, K.L.; Buettner, G.R.; et al. Ascorbate in pharmacologic concentrations selectively generates ascorbate radical and hydrogen peroxide in extracellular fluid in vivo. Proc. Natl. Acad. Sci. USA 2007, 104, 8749–8754.
- Sun, Y.; Pham, A.N.; Waite, T.D. The effect of vitamin C and iron on dopamine-mediated free radical generation: Implications to Parkinson’s disease. Dalton Trans 2018, 47, 4059–4069.
- Focarelli, F.; Giachino, A.; Waldron, K.J. Copper microenvironments in the human body define patterns of copper adaptation in pathogenic bacteria. PLoS Pathog. 2022, 18, e1010617.
- Tsang, T.; Davis, C.I.; Brady, D.C. Copper biology. Curr. Biol. 2021, 31, R421–R427.
- Tapiero, H.; Townsend, D.M.; Tew, K.D. Trace elements in human physiology and pathology. Copper. Biomed Pharm. 2003, 57, 386–398.
- Lutsenko, S.; Bhattacharjee, A.; Hubbard, A.L. Copper handling machinery of the brain. Metallomics 2010, 2, 596–608.
- Tarnacka, B.; Jopowicz, A.; Maślińska, M. Copper, Iron, and Manganese Toxicity in Neuropsychiatric Conditions. Int. J. Mol. Sci. 2021, 22, 7820.
- Amorós, R.; Murcia, M.; González, L.; Soler-Blasco, R.; Rebagliato, M.; Iñiguez, C.; Carrasco, P.; Vioque, J.; Broberg, K.; Levi, M.; et al. Maternal copper status and neuropsychological development in infants and preschool children. Int. J. Hyg. Environ. Health 2019, 222, 503–512.
- Gaier, E.D.; Eipper, B.A.; Mains, R.E. Copper signaling in the mammalian nervous system: Synaptic effects. J. Neurosci. Res. 2013, 91, 2–19.
- Gaetke, L.M.; Chow-Johnson, H.S.; Chow, C.K. Copper: Toxicological relevance and mechanisms. Arch. Toxicol. 2014, 88, 1929–1938.
- Da, S.; Rashed, L. Oxidative stress and genotoxicity among workers exposed to copper in a factory for non-ferrous industry in Egypt. Egypt. J. Occup. Med. 2019, 43, 1–15.
- Santos, S.; Silva, A.M.; Matos, M.; Monteiro, S.M.; Álvaro, A.R. Copper induced apoptosis in Caco-2 and Hep-G2 cells: Expression of caspases 3, 8 and 9, AIF and p53. Comp. Biochem. Physiol. Part C Toxicol. Pharm. 2016, 185–186, 138–146.
- Macip, S.; Igarashi, M.; Berggren, P.; Yu, J.; Lee, S.W.; Aaronson, S.A. Influence of induced reactive oxygen species in p53-mediated cell fate decisions. Mol. Cell. Biol. 2003, 23, 8576–8585.
- Narayanan, V.S.; Fitch, C.A.; Levenson, C.W. Tumor suppressor protein p53 mRNA and subcellular localization are altered by changes in cellular copper in human Hep G2 cells. J. Nutr. 2001, 131, 1427–1432.
- Gromadzka, G.; Tarnacka, B.; Flaga, A.; Adamczyk, A. Copper Dyshomeostasis in Neurodegenerative Diseases-Therapeutic Implications. Int. J. Mol. Sci. 2020, 21, 9259.
- Hozumi, I.; Hasegawa, T.; Honda, A.; Ozawa, K.; Hayashi, Y.; Hashimoto, K.; Yamada, M.; Koumura, A.; Sakurai, T.; Kimura, A.; et al. Patterns of levels of biological metals in CSF differ among neurodegenerative diseases. J. Neurol. Sci. 2011, 303, 95–99.
- Pall, H.S.; Williams, A.C.; Blake, D.R.; Lunec, J.; Gutteridge, J.M.; Hall, M.; Taylor, A. Raised cerebrospinal-fluid copper concentration in Parkinson’s disease. Lancet 1987, 2, 238–241.
- Genoud, S.; Senior, A.M.; Hare, D.J.; Double, K.L. Meta-Analysis of Copper and Iron in Parkinson’s Disease Brain and Biofluids. Mov. Disord. 2020, 35, 662–671.
- Santner, A.; Uversky, V.N. Metalloproteomics and metal toxicology of α-synuclein. Metallomics 2010, 2, 378–392.
- Gorell, J.M.; Johnson, C.C.; Rybicki, B.A.; Peterson, E.L.; Kortsha, G.X.; Brown, G.G.; Richardson, R.J. Occupational exposure to manganese, copper, lead, iron, mercury and zinc and the risk of Parkinson’s disease. Neurotoxicology 1999, 20, 239–247.
- Rybicki, B.A.; Johnson, C.C.; Uman, J.; Gorell, J.M. Parkinson’s disease mortality and the industrial use of heavy metals in Michigan. Mov. Disord. 1993, 8, 87–92.
- VanLandingham, J.W.; Tassabehji, N.M.; Somers, R.C.; Levenson, C.W. Expression profiling of p53-target genes in copper-mediated neuronal apoptosis. Neuromolecular Med. 2005, 7, 311–324.
- Lu, Y.; Prudent, M.; Fauvet, B.; Lashuel, H.A.; Girault, H.H. Phosphorylation of α-Synuclein at Y125 and S129 alters its metal binding properties: Implications for understanding the role of α-Synuclein in the pathogenesis of Parkinson’s Disease and related disorders. ACS Chem. Neurosci. 2011, 2, 667–675.
- Miotto, M.C.; Rodriguez, E.E.; Valiente-Gabioud, A.A.; Torres-Monserrat, V.; Binolfi, A.; Quintanar, L.; Zweckstetter, M.; Griesinger, C.; Fernández, C.O. Site-specific copper-catalyzed oxidation of α-synuclein: Tightening the link between metal binding and protein oxidative damage in Parkinson’s disease. Inorg. Chem. 2014, 53, 4350–4358.
- Anandhan, A.; Rodriguez-Rocha, H.; Bohovych, I.; Griggs, A.M.; Zavala-Flores, L.; Reyes-Reyes, E.M.; Seravalli, J.; Stanciu, L.A.; Lee, J.; Rochet, J.-C.; et al. Overexpression of alpha-synuclein at non-toxic levels increases dopaminergic cell death induced by copper exposure via modulation of protein degradation pathways. Neurobiol. Dis. 2015, 81, 76–92.
- Letelier, M.E.; FaúnNdez, M.; Jara-Sandoval, J.; Molina-Berrã os, A.; Cortãés-Troncoso, J.; Aracena-Parks, P.; Marín-Catalán, R. Mechanisms underlying the inhibition of the cytochrome P450 system by copper ions. J. Appl. Toxicol. 2009, 29, 695–702.
- Letelier, M.E.; Lepe, A.M.; Faúndez, M.; Salazar, J.; Marín, R.; Aracena, P.; Speisky, H. Possible mechanisms underlying copper-induced damage in biological membranes leading to cellular toxicity. Chem. Biol. Interact 2005, 151, 71–82.
- Kim, M.J.; Oh, S.B.; Kim, J.; Kim, K.; Ryu, H.S.; Kim, M.S.; Ayton, S.; Bush, A.I.; Lee, J.Y.; Chung, S.J. Association of metals with the risk and clinical characteristics of Parkinson’s disease. Park. Relat Disord 2018, 55, 117–121.
- Doguer, C.; Ha, J.H.; Collins, J.F. Intersection of Iron and Copper Metabolism in the Mammalian Intestine and Liver. Compr. Physiol. 2018, 8, 1433–1461.