Biometals are all metal ions that are essential for all living organisms. About 40% of all enzymes with known structures require biometals to function correctly. The main target of damage by biometals is the central nervous system (CNS). Biometal dysregulation (metal deficiency or overload) is related to pathological processes. Chronic occupational and environmental exposure to biometals, including iron and copper, is related to an increased risk of developing Parkinson’s disease (PD). Indeed, biometals have been shown to induce a dopaminergic neuronal loss in the substantia nigra. Although the etiology of PD is still unknown, oxidative stress dysregulation, mitochondrial dysfunction, and inhibition of both the ubiquitin–proteasome system (UPS) and autophagy are related to dopaminergic neuronal death.
- redox-active
- biometals
- iron
- copper
- Parkinson’s disease
12. Metals, Parkinson’s Disease, and Oxidative Stress
2. How Cells Die: Classical Mechanisms of Cell Death
Before discussing the role of biometals in neuronal death, it is worth summarizing the classical mechanisms of cell death. According to morphological, biochemical, and genetic characteristics, cell death is classified into three major types: apoptosis, necrosis, and autophagy. However, another section will discuss the latter, and additional cell death mechanisms will briefly be described. Apoptosis is well-characterized and known as programmed cell death type I (PCD type I). Its morphological changes include cell shrinkage, chromatin condensation (pyknosis), nuclear fragmentation (karyorrhexis), loss of plasma membrane integrity, and plasma membrane blebbing forming apoptotic bodies [38]. Necrosis, usually described as an accidental and uncontrollable mechanism, shows a substantial gain in cell volume (oncosis), swollen organelles, and disruption of the plasma membrane with the subsequent intracellular content release [39]. However, according to recent findings and following the guidelines of the Nomenclature Committee on Cell Death 2018, this classification has been updated, and mitochondrial permeability transition (MPT)-driven necrosis and necroptosis are now included, among other cell death subroutines [38]. MPT-driven necrosis is a regulated cell death induced by cell microenvironment disturbances that alter the inner mitochondrial membrane impermeability and is cyclophilin D (CYPD)-dependent [40,41,42]. Necroptosis is a regulated process activated by the recognition of extracellular and intracellular triggers through death receptors, and mixed lineage kinase domain-like pseudokinase (MLKL), receptor-interacting protein kinase 1 (RIPK1), and RIPK3 are crucial signaling molecules [43].3. Redox-Active Metals’ Role in Dopaminergic Neuronal Death
23.1. Iron
Iron is the most abundant metal on Earth, and almost all organisms have evolved to use this ubiquitous transition metal [25][45]. This metal is essential for the human body’s proper functioning. Iron is vital for oxygen transport (bound to hemoglobin), oxidative phosphorylation (bound to cytochrome C), neurotransmitter synthesis, myelin formation, and regulation of the biosynthesis of proteins such as ferritin and transferrin receptor (to store or mobilize iron) through iron-response proteins binding to iron-responsive elements at mRNA level [26][27][28][46,47,48]. In contrast, unbound iron causes cell toxicity as it can trigger a series of highly oxidative and toxic reactions [29][49]; this occurs when the iron concentrations exceed the binding capacity of transferrin [30][50]. Iron has a wide range of oxidative states, Fe2+ (ferrous) and Fe3+ (ferric) being the most common in biological environments [31][51]. Fe2+ binding to proteins is very unstable [32][52]. Iron’s neurotoxic effect has been related to the divalent metal ion transporter 1 (DMT1) overexpression, which imports iron into the cell, and can also be inhibited by H-ferritin [33][53]. Moreover, S-nitrosylation (SNO) of DMT1 cysteine thiol enhances Mn2+ and Fe2+ uptake [34][54]. Additionally, SNO-DMT1 has been detected in the post-mortem substantia nigra of PD patients [34][54]. The redox state of iron determines its role in cytotoxic reactions [35][55]. In a healthy brain, iron is distributed in a specific pattern by region and cell type. It is abundant in the substantia nigra and the basal ganglia, which are rich in dopaminergic neurons [36][56]. For over four decades, changes in iron and ferritin levels have been described in the brain of PD patients [15][27]. Several mechanisms are associated with iron-induced dopaminergic cell death, including (1) Fenton redox-reactions producing hydroxyl radicals [37][57]; (2) DA oxidative deamination, which is catalyzed by monoamine oxidase B (MAO-B) and regulated by Fe2+ and Fe3+ [38][39][58,59]; (3) 6-hydroxydopamine neurotoxic formation through DA metabolites reaction with iron and H2O2 [40][60]; and (4) increased rate of iron-induced α-synuclein fibril formation [21][33]. Iron takes part in the Fenton reaction producing free radicals; Fe2+ reacts with H2O2 or lipid peroxides to generate Fe3+, hydroxyl ion (OH–), and OH• or lipid radicals, which may lead to oxidative damage of macromolecules [27][41][47,61]. The Haber–Weiss reaction is where hydroxyl ion and hydroxyl radical are generated from the reaction of H2O2 and O2•– catalyzed by iron [42][62]. Additionally, the formation and accumulation of OH• lead to activation of the mitochondrial permeability transition pore (mPTP), which temporarily opens and increases ROS, provoking long-lasting activation and cell death. The latter is triggered by a decrease of ATP production, mitochondrial swelling, and rupture of the outer mitochondrial membrane, with subsequent release of mitochondrial death factors such as cytochrome C to the cytosolic compartment activating cell death by apoptosis [43][63]. Moreover, p53 is involved in mitochondrial dysfunction and oxidative stress mediated by Fe2+ in neuronal synaptic terminals [44][64]. Interestingly, lysosomes contain a redox-active iron pool derived from iron-rich macromolecules and cellular organelles, such as ferritin and mitochondria [45][46][65,66]. Most iron is found in a non-redox active form bound to ferritin. Ferritin degradation inside lysosomes during autophagy may be an intracellular redox-active iron source [47][67]. Next, H2O2 diffuses into lysosomes and reacts with the iron species through the Fenton and Fenton-like reactions, resulting in hydroxyl radical generation [45][65]. Intriguingly, defective mitochondria and lysosomes may promote receptor-interacting protein kinase 1 (RIPK1) activation, making cells susceptible to necroptosis [48][68]. Recently, necroptosis’s partial contribution to iron-mediated toxicity was demonstrated by using iron chelator deferoxamine (DFO) and the necroptosis inhibitor necrostatin 1 (NEC-1), significantly reducing cell death rates in the glutamate-induced model in vitro [49][69]. Iron accumulation in the brain is age-dependent, having the lowest levels at birth and a marked increase with age [50][70]. Brain regions associated with motor function have a high concentration of iron [50][70], and its levels in the whole brain are around 35.6–54.2 µg/g [51][71]. Its accumulation in the brain affects neurons. Exposure of neurons to iron induces oxidative stress, causing lipid peroxidation and DNA damage, which leads to caspase-dependent apoptotic cell death [52][72]. Several key mediators of ferroptosis have previously been implicated in PD pathogenesis. The SNpc is an iron-rich, dopamine (DA)-producing midbrain nucleus, which probably explains why it has a high risk of suffering neuronal death [53][54][73,74], mainly when iron accumulates, representing a PD feature [15][55][27,75]. Iron produces hydroxyl radicals with subsequent dopamine oxidation, likely contributing to an oxidative environment that increases the loss of nigral dopaminergic neurons in PD patients [56][76]. Moreover, genetic disorders that result in brain iron dyshomeostasis often cause Parkinsonism [57][58][59][77,78,79], demonstrating increased iron’s potential to contribute to PD pathogenesis. Indeed, mutations in several proteins involved in iron transport, increasing iron uptake and decreasing its export, are linked to PD. Mutant forms of transferrin, a critical protein for neuronal iron uptake, are associated with increased susceptibility to PD [60][61][80,81]. These data suggest that the iron uptake mechanism is overactive in these patients resulting in increased neuronal iron accumulation. Conversely, mutations in transferrin receptor 2 (TfR2) [61][81] are associated with a protective effect in PD, potentially due to reduced iron uptake. Neuronal iron export occurs via a transmembrane ion channel, ferroportin [62][82], and the Alzheimer’s disease (AD)-implicated amyloid precursor protein (APP) stabilizes ferroportin expression on the membrane to promote iron efflux [63][83]. In contrast, loss of APP membrane function results in impaired iron efflux and consequent neuronal iron retention [64][84]. Indeed, several rare variants of APP predispose individuals to PD, and several studies of familial AD indicate APP mutations are associated with Parkinsonism and LB formation [65][66][67][68][85,86,87,88]. Deficits in iron export in PD were further identified in the substantia nigra, with a significant depletion in APP expression levels independently of cell loss and an 80% decrease in ceruloplasmin (CP) activity [54][69][74,89]. Ceruloplasmin also has a ferrous oxidase activity and enables iron export by converting Fe2+ to Fe3+ [70][90], which is then bound to and removed by transferrin. Several point mutations in the CP-encoding gene are significantly associated with PD [71][91] and Parkinsonism [58][72][78,92], indicating that CP-mediated iron homeostasis is also likely involved in PD pathogenesis. Vitamin C, or ascorbic acid, has been shown to improve the absorption of Levodopa in some PD patients with poor Levodopa bioavailability [73][93]. However, it might be toxic as it loses one electron and forms an ascorbate radical (Asc•–). The electron can reduce metal ions such as iron and copper. The acidic extracellular environment favors the reduction of protein-centered metal, represented as Fe3+ reduction to Fe2+. Subsequently, Fe2+ donates an electron to O2 forming O2•– with subsequent dismutation to H2O2 [74][94]. Moreover, vitamin C administration may aggravate PD progression due to the possible peroxidation of Fe2+ bound to Asc•–. Therefore, combined vitamin C therapy for ROS scavenging and an iron chelator to sequester the metal may be a promising PD treatment option, reducing the toxicity induced by DA-derived quinones [75][95].(Figure 1)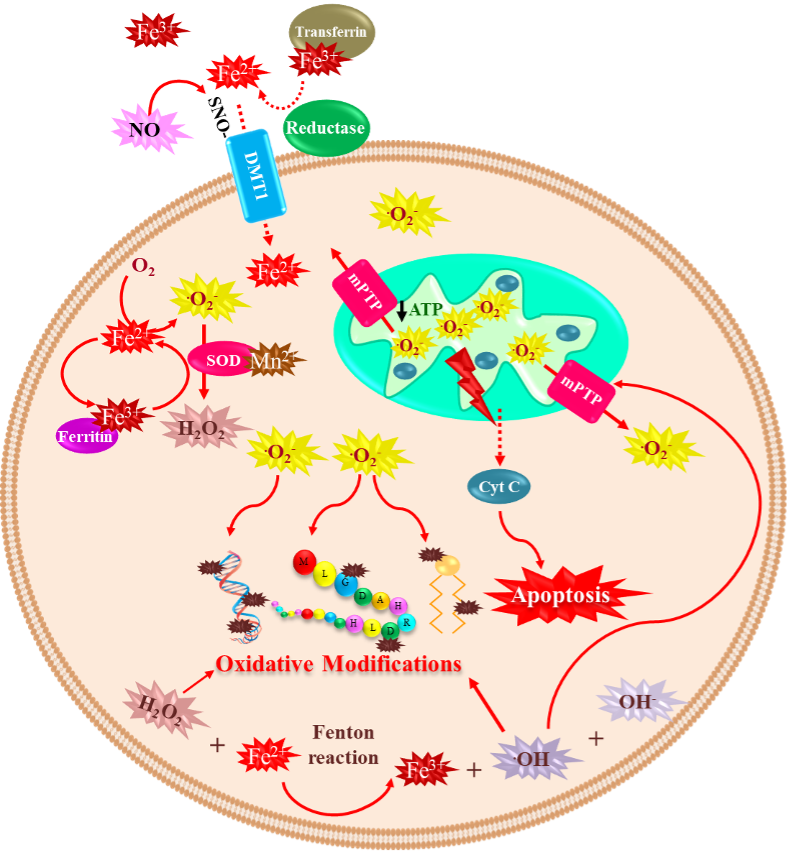
23.2. Copper
Copper is a trace element that constitutes 70 parts per million of the Earth’s crust. However, it is an essential micronutrient found in small amounts in tissues and cells, with a high concentration in the kidney, liver, and brain [76][111]. This metal functions as an essential cofactor and is required for structural and catalytic proprieties of more than 30 necessary enzymes; among them are ceruloplasmin, cytochrome oxidase, lysine oxidase, dopamine-hydroxylase, ascorbate oxidase, tyrosinase, and Cu/Zn SOD [77][112]. In living organisms, copper is mainly found oxidized (Cu2+) and reduced (Cu+) [78][113]. After the liver, the brain is the organ that accumulates the most significant amount of copper, reaching contents between 2.9 to 10 µg/g wet weight [79][114], and it is distributed differently in each region. The regions with the highest concentrations of copper are the substantia nigra, cerebellum, hippocampus, and hypothalamus [80][115]. Copper plays a crucial role in essential processes in CNS, such as brain development [81][116], antioxidant defense, synaptic transmission [82][117], and acting as an enzyme cofactor with oxidoreductase activity [77][112]. Notwithstanding, like any redox-active metal, copper becomes toxic when its intracellular accumulation is excessive, facilitating the formation of ROS and apoptotic processes [83][118]. When the cell is exposed to oxidative stress or copper, DNA damage and p53 expression are induced [84][85][119,120]. Furthermore, p53 undergoes oligomerization and phosphorylation to be translocated into the nucleus to induce genes such as BAX (BCL2 Associated X) and PUMA (p53 upregulated modulator of apoptosis) and subsequent release of cytochrome C into the cytosol to initiate apoptosis [86][87][121,122]. Therefore, copper plays a vital role in many diseases, such as Menkes disease, where copper is abnormally low in the brain. Conversely, in Wilson’s disease, the damage is caused by an excess of copper stored in brain tissue. Moreover, some neurodegenerative disorders such as AD, amyotrophic lateral sclerosis (ALS), prion disease, and PD have been linked to copper dyshomeostasis [88][123]. Increased copper levels have been reported in the cerebrospinal fluid and blood of PD patients [89][90][124,125]. However, a recent meta-analysis reported decreased copper levels in the substantia nigra of PD patients compared to healthy age-matched subjects [91][126]. Nevertheless, it has been demonstrated that chronic occupational exposure to copper increases the risk of developing PD [92][93][94][127,128,129]. Copper toxicity affects the basal ganglia and frontal cortex inducing Parkinson-like symptoms and cognitive deficits. The mechanism of copper-mediated toxicity includes cell cycle arrest via the upregulation of p21 (Cyclin Dependent Kinase Inhibitor 1A), reprimo (involved in regulating p53-dependent G2 arrest of the cell cycle and coded by RPRM gene), stathmin (microtubule destabilizing protein coded by STMN1 gene), and Tp53INP1 (Tumor Protein P53 Inducible Nuclear Protein 1). Additionally, stat-3 (Signal Transducer and Activator of Transcription 3), hsp70 (Heat Shock Protein 70), and hsp27 (Heat Shock Protein 27) are increased in an attempt to survive. Finally, p53-dependent and independent apoptosis are triggered, where IGFBP-6 (insulin-like growth factor binding protein-6), glutathione peroxidase, BCL-2, RB-1, PUMA, and several members of the redox-active PIG family of proteins, play a role [95][130]. Moreover, copper binding to α-synuclein increases oxidative stress and α-synuclein phosphorylation and accelerates the protein aggregation process [22][96][97][34,131,132]. In the neuroblastoma cell line SK-N-SH with dopaminergic phenotype, copper transporter protein 1 (Ctr1) overexpression led to intracellular glutathione depletion and potentiated the caspase-3-dependent-cell death induced by copper, indicating that copper’s toxicity is due to alterations in its intracellular homeostasis. In addition, copper-induced oxidative stress was primarily localized in the cytosol, and Nrf2 was upregulated to mediate an antioxidant response. In addition, copper increased protein ubiquitination, AMPK-Ulk1 signaling, p62, and Atg5-dependent autophagy as a protective mechanism [98][133]. The release of redox-active copper ions from copper-binding proteins and its binding to thiol or amine groups of cysteinyl and histidinyl residues of globular proteins, including enzymes, may result in conformational changes leading to its inactivation [99][100][134,135]. Paradoxically, some clinical trials report decreased circulating copper levels in PD patients compared to healthy controls [101][136], which may occur, because by binding to ceruloplasmin, copper stimulates ferroxidase activity and participates in iron homeostasis. Therefore, low levels of copper can indirectly generate toxicity by altering iron concentrations [102].(Figure 2)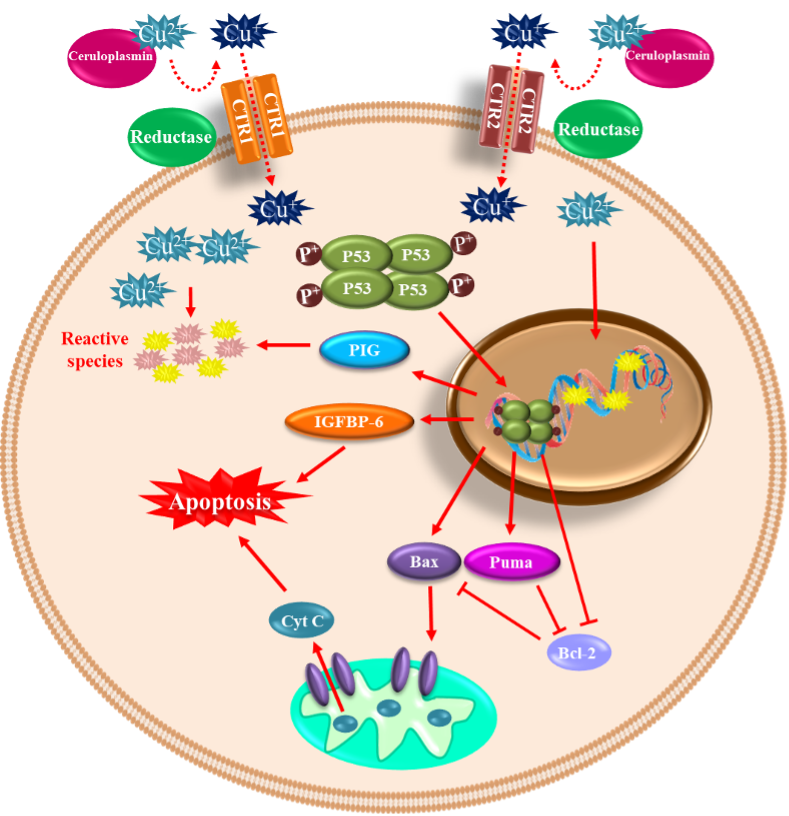 Figure 2. Copper-mediated neuronal cell death. Copper can be found in its oxidized (Cu2+) and reduced (Cu+) forms within the cells. The oxidized form of copper is bound to proteins, and when reduced by a reductase enzyme, it enters the cell through the transporters CTR1 and CTR2. Once inside and in high concentration, it induces DNA damage and p53 expression. p53 undergoes oligomerization/phosphorylation and is translocated into the nucleus to induce BAX and PUMA, with the consequent release of cytochrome C into the cytosol to initiate apoptosis. p53-dependent and independent apoptosis is triggered, where insulin-like growth factor binding protein-6 (IGFBP-6) and PIG proteins (prooxidant proteins), among other players, are involved.
In summary, nearly 40% of the proteins need a biometal as a cofactor to fulfill their function. Therefore, it is crucial to understand the transition metals’ role in health and disease because their dyshomeostasis (deficiency or overloading) is closely related to different disorders and mainly to neurodegeneration. Research in this field has found that biometals are tightly regulated because a narrow unbalance provokes diseases such as PD. Therefore, understanding the complexity of the interaction between transition metals and proteins can shed light on possible neurodegeneration biomarkers for preventing neuronal cell death. Hopefully, neurodegeneration may be achieved by either supplementing transition metals when there is a deficiency or using chelating agents to avoid metal overload that induces neuronal cell death in PD[137].
Figure 2. Copper-mediated neuronal cell death. Copper can be found in its oxidized (Cu2+) and reduced (Cu+) forms within the cells. The oxidized form of copper is bound to proteins, and when reduced by a reductase enzyme, it enters the cell through the transporters CTR1 and CTR2. Once inside and in high concentration, it induces DNA damage and p53 expression. p53 undergoes oligomerization/phosphorylation and is translocated into the nucleus to induce BAX and PUMA, with the consequent release of cytochrome C into the cytosol to initiate apoptosis. p53-dependent and independent apoptosis is triggered, where insulin-like growth factor binding protein-6 (IGFBP-6) and PIG proteins (prooxidant proteins), among other players, are involved.
In summary, nearly 40% of the proteins need a biometal as a cofactor to fulfill their function. Therefore, it is crucial to understand the transition metals’ role in health and disease because their dyshomeostasis (deficiency or overloading) is closely related to different disorders and mainly to neurodegeneration. Research in this field has found that biometals are tightly regulated because a narrow unbalance provokes diseases such as PD. Therefore, understanding the complexity of the interaction between transition metals and proteins can shed light on possible neurodegeneration biomarkers for preventing neuronal cell death. Hopefully, neurodegeneration may be achieved by either supplementing transition metals when there is a deficiency or using chelating agents to avoid metal overload that induces neuronal cell death in PD[137].