Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Kin Wah Terence Lee | -- | 1879 | 2023-01-17 07:54:21 | | | |
| 2 | Catherine Yang | Meta information modification | 1879 | 2023-01-17 08:40:09 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Leung, R.W.H.; Lee, T.K.W. Wnt/β-Catenin Signaling Pathway in Hepatocellular Carcinoma. Encyclopedia. Available online: https://encyclopedia.pub/entry/40264 (accessed on 24 July 2026).
Leung RWH, Lee TKW. Wnt/β-Catenin Signaling Pathway in Hepatocellular Carcinoma. Encyclopedia. Available at: https://encyclopedia.pub/entry/40264. Accessed July 24, 2026.
Leung, Rainbow Wing Hei, Terence Kin Wah Lee. "Wnt/β-Catenin Signaling Pathway in Hepatocellular Carcinoma" Encyclopedia, https://encyclopedia.pub/entry/40264 (accessed July 24, 2026).
Leung, R.W.H., & Lee, T.K.W. (2023, January 17). Wnt/β-Catenin Signaling Pathway in Hepatocellular Carcinoma. In Encyclopedia. https://encyclopedia.pub/entry/40264
Leung, Rainbow Wing Hei and Terence Kin Wah Lee. "Wnt/β-Catenin Signaling Pathway in Hepatocellular Carcinoma." Encyclopedia. Web. 17 January, 2023.
Copy Citation
Hepatocellular carcinoma (HCC) is a major cause of cancer death worldwide due to its high rates of tumor recurrence and metastasis. Aberrant Wnt/β-catenin signaling has been shown to play a significant role in HCC development, progression and clinical impact on tumor behavior.
cancer metabolism
drug resistance
metabolic reprogramming
hepatocellular carcinoma
Wnt/β-catenin
1. Mutation and Expression Status of the Wnt/β-Catenin Pathway and Its Clinical Significance
Wnt/β-catenin signaling is crucial in contributing to HCC pathogenesis, where genetic mutations and epigenetic alterations are primarily revealed [1]. Activation of the Wnt/β-catenin signaling pathway was discovered in 20–35% of HCC cases, among which most are resulted by gene mutations of the key genes, including CTNNB1, AXIN, and APC [2][3][4]. In this case, CTNNB1 is the gene that specifically encodes β-catenin. Mutation of β-catenin is positively related to HCC progression due to its oncogenic role [5]. To date, mutations at the serine/threonine sites of exon 3 of the β-catenin gene are mostly found to be involved in the phosphorylation and ubiquitination of β-catenin, thus enhancing its nuclear translocation in approximately 20% of HCC cases [6][7]. In addition, conventional and missense mutations have also been reported in other codons of β-catenin [8]. Previous reports showed that conventional mutations at codons 33, 37, 41, and 45 are discovered in over 12% of HCC patients, where missense mutations are observed at codons 32, 34, and 35 [8], which indicates the capability of mutated β-catenin proteins to evade degradation and enter the nucleus [6][9]. It is also noted that tumor cells with aberrant Wnt/β-catenin activation due to the mutation of β-catenin that tend to grow and spread more quickly in HCC [5].
Apart from β-catenin, deregulation of the Wnt/β-catenin signaling pathway is also caused by mutations in protein degradation complexes [10]. These mutations cause dysfunction of the destruction complex and accumulation of β-catenin in the nucleus in approximately 40–70% of HCC cases [1]. One example is the amino acid substitution in armadillo repeats domain 5/6 of β-catenin in human HCC cases [11]. This results in a reduction of APC binding to the degradation complex, which activates the Wnt/β-catenin signaling pathway and enhances targeted gene transcription [11]. It has been reported that a small amount of β-catenin accumulated in the nucleus is sufficient to activate Wnt target genes, suggesting the crucial role of β-catenin in HCC progression [12][13]. However, several studies have shown that the mutation of β-catenin alone is insufficient for promoting HCC in mice, which is different in comparison with humans [13][14], as the tumorigenic potential could be augmented when combined with other oncogenic pathways, such as H-RAS, MET, AKT, or chemicals such as diethylnitrosamine (DEN) [1][15].
In addition, high levels of E-cadherin have been reported to be correlated with the accumulation of β-catenin in both the cytosol and nucleus, that drives the transcription of Wnt target genes [16]. C-Myc and cyclin D, as key Wnt-target genes, not only perform their roles as proto-oncogenes for tumor formation but also regulate liver cancer stem cell (CSC) properties by mediating various signaling pathways involved in cellular differentiation and survival [5][6]. As previously mentioned, HBV and HCV are the causes of HCC, in which they lead to genetic mutations in genes involved in Wnt/β-catenin signaling [16][17]. It is common to find CTNNB1 mutations in HCV-related HCC rather than HBV-related HCC or nonviral HCC [10]. However, mutation of Axin1 is more often found in HBV-related HCC tumors [10]. Interestingly, apart from the mutations of the canonical pathway, Zucman-Rossi et al. suggested that Axin1 mutation also plays a role in exerting oncogenic effects manifested by overexpressing glutamine synthase (GS), leading to β-catenin activation that correlates to the non-canonical pathway [18].
2. Regulation of Wnt/β-Catenin Pathway in HCC
2.1. Epigenetic Regulation of Wnt/β-Catenin
Several epigenetic dysregulations contribute to Wnt/β-catenin activation in HCC. DNA methylation is crucial in maintaining CSC properties, in which its inhibition can influence the fate of cells and gene expressions [19]. For instance, DNA methyltransferase (DNMT) plays a role in catalyzing the transition between a methyl group and DNA, mediating BEX1 expression in HCC [20]. A decrease in DNMT1 results in BEX1 hypomethylation that further enhances the transcription of β-catenin, which causes the activation of the Wnt/β-catenin signaling pathway [20]. Moreover, secreted frizzled-related proteins (SFRPs) negatively regulate Wnt/β-catenin signaling via DNA methylation, representing a leading cause of activating β-catenin activity in HCC [21]. Another study also consistently showed that downregulation of the SFRP family is correlated with Wnt/β-catenin signaling activation, in which SFRP1 and SFRP5 are also found to enhance the progression of HCC [22][23]. Similarly, downregulating Wnt inhibitory factor 1 (WIF1) or Dickkopf-related protein 3 (DKK3) has been proven to result in common consequences for SFRPs [24]. In addition, SOX17 is reported to take part in the aberrant activation of Wnt/β-catenin signaling due to promoter methylation [25]. Silencing of SOX17 could enhance Wnt activity due to the failure in interacting with TCF/LEF, which hinders Wnt-target gene transcription [26]. Apart from the genomic instability caused by DNA hypomethylation, another study showed the involvement of potassium channels in epigenetic regulation of the Wnt/β-catenin pathway [27]. Fan et al. revealed that a decrease in KCNQ1 (potassium voltage-gated channel subfamily Q member 1) causes an increment in Wnt/β-catenin activity via DNA hypermethylation [27].
Furthermore, several alterations through histone modification have been reported in HCC. Enhancer of zeste homologous 2 (EZH2) is a histone methyltransferase that plays a role in catalyzing methylation of histone H3 to achieve repression of Wnt antagonists, promoting Wnt/β-catenin signaling and hepatocarcinogenesis [28][29]. Histone deacetylases (HDAC) have been revealed to interact with EZH2 through its enzymatic role [30]. Specifically, for HDAC8, its upregulation due to the chromatin modifications is coexpressed with the lipogenic transcription factor SREBP1 in HCC mouse models, causing cell cycle arrest and β-catenin activation, which drives NAFLD-induced hepatocarcinogenesis [30]. Moreover, HDAC8 can also bind to pyruvate kinase M2 (PKM2) and subsequently deacetylate the residue K62, prompting the nuclear translocation of PKM2 and the binding of β-catenin that results in Wnt target gene transcription [31]. Similarly, EZH2 overexpression elevated the levels of the oncogene H3K27me3, which silenced Wnt inhibitors, leading to induced cell proliferation with activated β-catenin activity [28].
2.2. Non-Coding RNAs in Regulation of Wnt/β-Catenin
It has been suggested that microRNAs (miRNAs) and long noncoding RNAs (lncRNAs) are critical regulators associated with various tumors, in which they are negatively regulated [32]. Dysregulation of miRNAs and lncRNAs could lead to tumorigenesis in HCC. LncRNA-miRNA binding yields a complete endogenous RNA (ceRNA) that can avoid messenger RNA (mRNA) recognition and further silencing effects, known as the “sponge effect” [32]. Mounting evidence suggests that miRNA sponges are involved in Wnt/β-catenin signaling and are associated with HCC progression (Table 1). For example, LINC00355 and LINC01278 are negative regulators of miR-217-5p and miR-1258, respectively [33][34]. Overexpression of lncRNAs downregulate the corresponding miRNAs and further activates Wnt/β-catenin signaling, resulting in increased levels of Wnt target gene transcription and metastatic ability of HCC cells [33][34]. Additionally, upregulation of LINC00662 in HCC induced WNT3A secretion with miR-15a/16/107 binding, resulting in the activation of Wnt/β-catenin and polarizes M2 macrophage [35]. Similarly, overexpressing FEZF1-AS1 negatively regulates the level of miR-107, which inhibits the activation of Wnt/β-catenin signaling, while downregulation of FEZF1-AS1 enhances the expression of β-catenin [36]. Furthermore, both miR-122 and miR-148a were found to contribute to liver cancers by binding to the 3′-untranslated region (3′-UTR) site of Wnt1, suppressing the level of β-catenin and inhibiting Wnt-target gene transcription [37][38]. Furthermore, a decrease in these miRNA levels could cause excess Wnt/β-catenin signaling and increase EMT [37]. All the above mentioned enhance the progression of HCC. As a tumor suppressor, miR-34a is reported in mice and HCC patients and found to be upregulated through activated Wnt/β-catenin signaling [39]. In addition, overexpression of miR-145 has been shown to diminish the level of β-catenin, suppressing HCC cell growth [40]. To sum up, Wnt/β-catenin signaling is tightly regulated by DNA methylation, histone modification and non-coding RNAs in HCC (Figure 1).
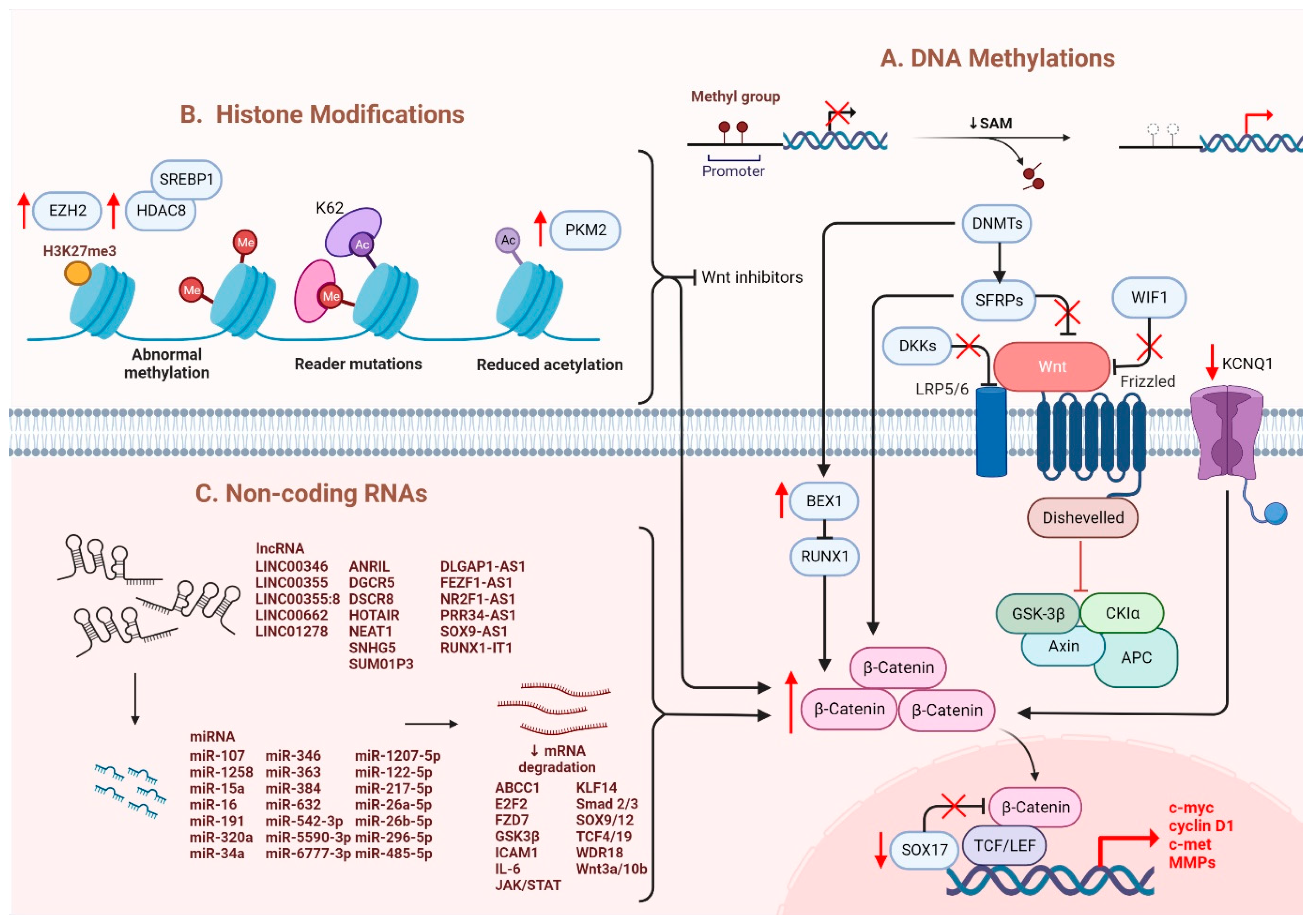
Figure 1. Regulation of Wnt/β-catenin signaling in HCC. Wnt/β-catenin signaling in HCC is regulated by (A) DNA methylation, (B) histone modification and (C) non-coding RNAs.
Table 1. The list of lncRNAs and their related miRNAs in regulation of Wnt/β-catenin signaling in HCC.
| LncRNA | miRNA | Targets | Ref. |
|---|---|---|---|
| LINC00346 | miR-542-3p | FZD7, WDR18 | [41] |
| LINC00355 | miR-217-5p | GSK3β, c-myc, CCND1 | [33] |
| LINC00355:8 | miR-6777-3p | Wnt10b | [42] |
| LINC00662 | miR-15a, miR-16, miR-107 | Wnt3a | [35] |
| LINC01278 | miR-1258 | TCF-4, Smad2/3 | [34] |
| ANRIL | miR-191, miR-122-5p |
CCND1, p53, p21, MMP-2, MMP-9, Vimentin | [43][44] |
| DGCR5 | miR-346 | KLF14 | [45] |
| DSCR8 | miR-485-5p | FZD7 | [46] |
| DLGAP1-AS1 | miR-26a-5p, miR-26b-5p |
IL-6, JAK2, STAT3 | [47] |
| FEZF1-AS1 | miR-107 | Wnt3a, ICAM1, Vimentin | [36] |
| HOTAIR | miR-34a | Akt | [48] |
| MIR194-2HG | miR-1207-5p | TCF19 | [49] |
| NEAT1 | miR-384 | Wnt | [50] |
| NR2F1-AS1 | miR-363 | ABCC1 | [51] |
| PRR34-AS1 | miR-296-5p | E2F2, SOX12 | [52] |
| RUNX1-IT1 | miR-632 | GSK3β | [53] |
| SNHG5 | miR-26a-5p | GSK3β | [54] |
| SUMO1P3 | miR-320a | C-myc, CCND1 | [55] |
| SOX9-AS1 | miR-5590-3p | SOX9 | [56] |
2.3. Other Molecules Involved in the Regulation of Wnt/β-Catenin
Apart from genetic mutations and epigenetic dysregulation, other molecules/pathways were identified to regulate Wnt/β-catenin signaling. In normoric environment, ROS is maintained at a low level; whereas a steady increase of ROS level promotes cancer development and progression [57]. A recent study showed that Wnt/β-catenin signaling was suppressed upon elevation of intracellular ROS level [5][58]. In HCC, glutaminase 1 (GLS1) is upregulated which augmented liver CSC properties with increased expression of CSC markers via suppression of ROS level [5][58]. Likewise, another study also showed that ROS accumulation due to the overexpression of Cytochrome P450 2E1 (CYP2E1) decreased the activity of Wnt/β-catenin signaling through the degradation of DVL2 in HCC [59]. Hypoxia also plays a crucial role in the activation of Wnt/β-catenin signaling. Hypoxia-inducible factor 1-alpha (HIF1α), a hypoxia-inducible factor, regulates transcription in hypoxic environments and is also reported to mediate the expression of B-cell lymphoma 9 (BCL9) [16][60]. BCL9 can coactivate with HIF1α to enhance the transcriptional activity of β-catenin regardless of whether genetic mutations occur, resulting in activation of Wnt/β-catenin signaling and leading to HCC progression [60]. Furthermore, ZBTB20 has been reported in liver tumorigenesis with its role in suppressing PPARG expression and inhibiting proteasomal degradation of the β-catenin destruction complex [61]. Overall, once the nuclear translocation of β-catenin is achieved, the expression levels of the downstream genes involved in EMT are modulated and enhanced, causing hepatocarcinogenesis [3]. C-Myc is the most critical gene induced by activated Wnt/β-catenin signaling, which enhances the mechanisms of glycolysis and glutaminolysis [62]. This is followed by cyclin D1, which has been reported to be enhanced in both mouse and human HCC [63][64]. Specifically, overexpression of c-Met and cyclin D1 triggers the development of liver tumors and decreases survival in mice [65]. It is also noted that upregulation of cyclin D1 enhances tumor metastatic ability [66]. Additionally, studies have discovered that GS and VEGF are also involved in modulating the downstream effects of activated Wnt and assisting in angiogenesis [67], as the upregulation of multiple matrix metalloproteinases (MMPs), including MMP2 and MMP9, is associated with tumor metastasis [68]. Apart from gene regulation, aberrant β-catenin signaling also negatively regulates certain signaling cascades: for example, the suppression of NF-κB cascade in the liver [69]. Moreover, the crosstalk between Wnt and Hippo signaling pathways has been observed in HCC. Recent study showed that Wnt-Hippo signature related genes may be a potential markers for prediction of immune infiltration in HCC [70]. Notably, aberrant activation of β-catenin caused by the deletion of mammalian STE20-like protein kinase 1/2 (Mst1/2) promotes tumor growth, indicating the co-expression of YAP and β-catenin in HCC [71].
References
- He, S.; Tang, S. WNT/β-catenin signaling in the development of liver cancers. Biomed. Pharmacother. 2020, 132, 110851.
- Russell, J.O.; Monga, S.P. Wnt/β-Catenin Signaling in Liver Development, Homeostasis, and Pathobiology. Annu. Rev. Pathol. 2018, 13, 351–378.
- Xu, C.; Xu, Z.; Zhang, Y.; Evert, M.; Calvisi, D.F.; Chen, X. β-Catenin signaling in hepatocellular carcinoma. J. Clin. Investig. 2022, 132, e154515.
- Lu, L.C.; Shao, Y.Y.; Lee, Y.H.; Hsieh, M.S.; Hsiao, C.H.; Lin, H.H.; Kao, H.F.; Ma, Y.Y.; Yen, F.C.; Cheng, A.L.; et al. β-catenin (CTNNB1) mutations are not associated with prognosis in advanced hepatocellular carcinoma. Oncology 2014, 87, 159–166.
- Deldar Abad Paskeh, M.; Mirzaei, S.; Ashrafizadeh, M.; Zarrabi, A.; Sethi, G. Wnt/β-Catenin Signaling as a Driver of Hepatocellular Carcinoma Progression: An Emphasis on Molecular Pathways. J. Hepatocell. Carcinoma 2021, 8, 1415–1444.
- Monga, S.P. β-Catenin Signaling and Roles in Liver Homeostasis, Injury, and Tumorigenesis. Gastroenterology 2015, 148, 1294–1310.
- Javanmard, D.; Najafi, M.; Babaei, M.R.; Karbalaie Niya, M.H.; Esghaei, M.; Panahi, M.; Safarnezhad Tameshkel, F.; Tavakoli, A.; Jazayeri, S.M.; Ghaffari, H.; et al. Investigation of CTNNB1 gene mutations and expression in hepatocellular carcinoma and cirrhosis in association with hepatitis B virus infection. Infect. Agents Cancer 2020, 15, 37.
- Okabe, H.; Kinoshita, H.; Imai, K.; Nakagawa, S.; Higashi, T.; Arima, K.; Uchiyama, H.; Ikegami, T.; Harimoto, N.; Itoh, S.; et al. Diverse Basis of β-Catenin Activation in Human Hepatocellular Carcinoma: Implications in Biology and Prognosis. PLoS ONE 2016, 11, e0152695.
- Wong, C.M.; Fan, S.T.; Ng, I.O. beta-Catenin mutation and overexpression in hepatocellular carcinoma: Clinicopathologic and prognostic significance. Cancer 2001, 92, 136–145.
- Wang, W.; Pan, Q.; Fuhler, G.M.; Smits, R.; Peppelenbosch, M.P. Action and function of Wnt/β-catenin signaling in the progression from chronic hepatitis C to hepatocellular carcinoma. J. Gastroenterol. 2017, 52, 419–431.
- Liu, P.; Liang, B.; Liu, M.; Lebbink, J.H.G.; Li, S.; Qian, M.; Lavrijsen, M.; Peppelenbosch, M.P.; Chen, X.; Smits, R. Oncogenic Mutations in Armadillo Repeats 5 and 6 of β-Catenin Reduce Binding to APC, Increasing Signaling and Transcription of Target Genes. Gastroenterology 2020, 158, 1029–1043.e10.
- Kim, E.; Lisby, A.; Ma, C.; Lo, N.; Ehmer, U.; Hayer, K.E.; Furth, E.E.; Viatour, P. Promotion of growth factor signaling as a critical function of β-catenin during HCC progression. Nat. Commun. 2019, 10, 1909.
- Kim, S.; Jeong, S. Mutation Hotspots in the β-Catenin Gene: Lessons from the Human Cancer Genome Databases. Mol. Cells 2019, 42, 8–16.
- Harada, N.; Miyoshi, H.; Murai, N.; Oshima, H.; Tamai, Y.; Oshima, M.; Taketo, M.M. Lack of tumorigenesis in the mouse liver after adenovirus-mediated expression of a dominant stable mutant of beta-catenin. Cancer Res. 2002, 62, 1971–1977.
- Liu, L.J.; Xie, S.X.; Chen, Y.T.; Xue, J.L.; Zhang, C.J.; Zhu, F. Aberrant regulation of Wnt signaling in hepatocellular carcinoma. World J. Gastroenterol. 2016, 22, 7486–7499.
- Khalaf, A.M.; Fuentes, D.; Morshid, A.I.; Burke, M.R.; Kaseb, A.O.; Hassan, M.; Hazle, J.D.; Elsayes, K.M. Role of Wnt/β-catenin signaling in hepatocellular carcinoma, pathogenesis, and clinical significance. J. Hepatocell. Carcinoma 2018, 5, 61–73.
- Inagawa, S.; Itabashi, M.; Adachi, S.; Kawamoto, T.; Hori, M.; Shimazaki, J.; Yoshimi, F.; Fukao, K. Expression and prognostic roles of beta-catenin in hepatocellular carcinoma: Correlation with tumor progression and postoperative survival. Clin. Cancer Res. 2002, 8, 450–456.
- Zucman-Rossi, J.; Benhamouche, S.; Godard, C.; Boyault, S.; Grimber, G.; Balabaud, C.; Cunha, A.S.; Bioulac-Sage, P.; Perret, C. Differential effects of inactivated Axin1 and activated beta-catenin mutations in human hepatocellular carcinomas. Oncogene 2007, 26, 774–780.
- Raggi, C.; Factor, V.M.; Seo, D.; Holczbauer, A.; Gillen, M.C.; Marquardt, J.U.; Andersen, J.B.; Durkin, M.; Thorgeirsson, S.S. Epigenetic reprogramming modulates malignant properties of human liver cancer. Hepatology 2014, 59, 2251–2262.
- Wang, Q.; Liang, N.; Yang, T.; Li, Y.; Li, J.; Huang, Q.; Wu, C.; Sun, L.; Zhou, X.; Cheng, X.; et al. DNMT1-mediated methylation of BEX1 regulates stemness and tumorigenicity in liver cancer. J. Hepatol. 2021, 75, 1142–1153.
- Lin, Y.W.; Shih, Y.L.; Lien, G.S.; Suk, F.M.; Hsieh, C.B.; Yan, M.D. Promoter methylation of SFRP3 is frequent in hepatocellular carcinoma. Dis. Markers 2014, 2014, 351863.
- Xie, Q.; Chen, L.; Shan, X.; Shan, X.; Tang, J.; Zhou, F.; Chen, Q.; Quan, H.; Nie, D.; Zhang, W.; et al. Epigenetic silencing of SFRP1 and SFRP5 by hepatitis B virus X protein enhances hepatoma cell tumorigenicity through Wnt signaling pathway. Int. J. Cancer 2014, 135, 635–646.
- Takagi, H.; Sasaki, S.; Suzuki, H.; Toyota, M.; Maruyama, R.; Nojima, M.; Yamamoto, H.; Omata, M.; Tokino, T.; Imai, K.; et al. Frequent epigenetic inactivation of SFRP genes in hepatocellular carcinoma. J. Gastroenterol. 2008, 43, 378–389.
- Ding, Z.; Qian, Y.B.; Zhu, L.X.; Xiong, Q.R. Promoter methylation and mRNA expression of DKK-3 and WIF-1 in hepatocellular carcinoma. World J. Gastroenterol. 2009, 15, 2595–2601.
- Jia, Y.; Yang, Y.; Liu, S.; Herman, J.G.; Lu, F.; Guo, M. SOX17 antagonizes WNT/β-catenin signaling pathway in hepatocellular carcinoma. Epigenetics 2010, 5, 743–749.
- Tian, Y.; Mok, M.T.; Yang, P.; Cheng, A.S. Epigenetic Activation of Wnt/β-Catenin Signaling in NAFLD-Associated Hepatocarcinogenesis. Cancers 2016, 8, 76.
- Fan, H.; Zhang, M.; Liu, W. Hypermethylated KCNQ1 acts as a tumor suppressor in hepatocellular carcinoma. Biochem. Biophys. Res. Commun. 2018, 503, 3100–3107.
- Song, H.; Yu, Z.; Sun, X.; Feng, J.; Yu, Q.; Khan, H.; Zhu, X.; Huang, L.; Li, M.; Mok, M.T.S.; et al. Androgen receptor drives hepatocellular carcinogenesis by activating enhancer of zeste homolog 2-mediated Wnt/β-catenin signaling. EBioMedicine 2018, 35, 155–166.
- Cheng, A.S.; Lau, S.S.; Chen, Y.; Kondo, Y.; Li, M.S.; Feng, H.; Ching, A.K.; Cheung, K.F.; Wong, H.K.; Tong, J.H.; et al. EZH2-mediated concordant repression of Wnt antagonists promotes β-catenin-dependent hepatocarcinogenesis. Cancer Res. 2011, 71, 4028–4039.
- Tian, Y.; Wong, V.W.; Wong, G.L.; Yang, W.; Sun, H.; Shen, J.; Tong, J.H.; Go, M.Y.; Cheung, Y.S.; Lai, P.B.; et al. Histone Deacetylase HDAC8 Promotes Insulin Resistance and β-Catenin Activation in NAFLD-Associated Hepatocellular Carcinoma. Cancer Res. 2015, 75, 4803–4816.
- Zhang, R.; Shen, M.; Wu, C.; Chen, Y.; Lu, J.; Li, J.; Zhao, L.; Meng, H.; Zhou, X.; Huang, G.; et al. HDAC8-dependent deacetylation of PKM2 directs nuclear localization and glycolysis to promote proliferation in hepatocellular carcinoma. Cell Death Dis. 2020, 11, 1036.
- Shi, T.; Morishita, A.; Kobara, H.; Masaki, T. The Role of Long Non-Coding RNA and microRNA Networks in Hepatocellular Carcinoma and Its Tumor Microenvironment. Int. J. Mol. Sci. 2021, 22, 630.
- Luo, X.; ABudureyimu, M.; Yang, G.; Yan, Z.; Fu, X.; Lu, P.; Zhang, D.; Zhang, S.; Ding, Z. LINC00355 triggers malignant progression of hepatocellular carcinoma via the sponge effect on miR-217-5p with the involvement of the Wnt/β-catenin signaling. J. BUON 2021, 26, 1964–1969.
- Huang, W.J.; Tian, X.P.; Bi, S.X.; Zhang, S.R.; He, T.S.; Song, L.Y.; Yun, J.P.; Zhou, Z.G.; Yu, R.M.; Li, M. The β-catenin/TCF-4-LINC01278-miR-1258-Smad2/3 axis promotes hepatocellular carcinoma metastasis. Oncogene 2020, 39, 4538–4550.
- Tian, X.; Wu, Y.; Yang, Y.; Wang, J.; Niu, M.; Gao, S.; Qin, T.; Bao, D. Long noncoding RNA LINC00662 promotes M2 macrophage polarization and hepatocellular carcinoma progression via activating Wnt/β-catenin signaling. Mol. Oncol. 2020, 14, 462–483.
- Yao, J.; Yang, Z.; Yang, J.; Wang, Z.G.; Zhang, Z.Y. Long non-coding RNA FEZF1-AS1 promotes the proliferation and metastasis of hepatocellular carcinoma via targeting miR-107/Wnt/β-catenin axis. Aging 2021, 13, 13726–13738.
- Yan, H.; Dong, X.; Zhong, X.; Ye, J.; Zhou, Y.; Yang, X.; Shen, J.; Zhang, J. Inhibitions of epithelial to mesenchymal transition and cancer stem cells-like properties are involved in miR-148a-mediated anti-metastasis of hepatocellular carcinoma. Mol. Carcinog. 2014, 53, 960–969.
- Xu, J.; Zhu, X.; Wu, L.; Yang, R.; Yang, Z.; Wang, Q.; Wu, F. MicroRNA-122 suppresses cell proliferation and induces cell apoptosis in hepatocellular carcinoma by directly targeting Wnt/β-catenin pathway. Liver Int. 2012, 32, 752–760.
- Gougelet, A.; Sartor, C.; Bachelot, L.; Godard, C.; Marchiol, C.; Renault, G.; Tores, F.; Nitschke, P.; Cavard, C.; Terris, B.; et al. Antitumour activity of an inhibitor of miR-34a in liver cancer with β-catenin-mutations. Gut 2016, 65, 1024–1034.
- Jin, X.; Chen, Y.P.; Kong, M.; Zheng, L.; Yang, Y.D.; Li, Y.M. Transition from hepatic steatosis to steatohepatitis: Unique microRNA patterns and potential downstream functions and pathways. J. Gastroenterol. Hepatol. 2012, 27, 331–340.
- Zhang, N.; Chen, X. A positive feedback loop involving the LINC00346/β-catenin/MYC axis promotes hepatocellular carcinoma development. Cell Oncol. 2020, 43, 137–153.
- Zhou, F.; Lei, Y.; Xu, X.; Zhou, H.; Liu, H.; Jiang, J.; Yang, Y.; Wu, B. LINC00355:8 promotes cell proliferation and migration with invasion via the MiR-6777-3p/Wnt10b axis in Hepatocellular Carcinoma. J. Cancer 2020, 11, 5641–5655.
- Huang, D.; Bi, C.; Zhao, Q.; Ding, X.; Bian, C.; Wang, H.; Wang, T.; Liu, H. Knockdown long non-coding RNA ANRIL inhibits proliferation, migration and invasion of HepG2 cells by down-regulation of miR-191. BMC Cancer 2018, 18, 919.
- Ma, J.; Li, T.; Han, X.; Yuan, H. Knockdown of LncRNA ANRIL suppresses cell proliferation, metastasis, and invasion via regulating miR-122-5p expression in hepatocellular carcinoma. J. Cancer Res. Clin. Oncol. 2018, 144, 205–214.
- Wang, Y.G.; Liu, J.; Shi, M.; Chen, F.X. LncRNA DGCR5 represses the development of hepatocellular carcinoma by targeting the miR-346/KLF14 axis. J. Cell. Physiol. 2018, 234, 572–580.
- Wang, Y.; Sun, L.; Wang, L.; Liu, Z.; Li, Q.; Yao, B.; Wang, C.; Chen, T.; Tu, K.; Liu, Q. Long non-coding RNA DSCR8 acts as a molecular sponge for miR-485-5p to activate Wnt/β-catenin signal pathway in hepatocellular carcinoma. Cell Death Dis. 2018, 9, 851.
- Lin, Y.; Jian, Z.; Jin, H.; Wei, X.; Zou, X.; Guan, R.; Huang, J. Long non-coding RNA DLGAP1-AS1 facilitates tumorigenesis and epithelial-mesenchymal transition in hepatocellular carcinoma via the feedback loop of miR-26a/b-5p/IL-6/JAK2/STAT3 and Wnt/β-catenin pathway. Cell Death Dis. 2020, 11, 34.
- Duan, Y.; Chen, J.; Yang, Y.; Qu, Z.; Lu, Y.; Sun, D. LncRNA HOTAIR contributes Taxol-resistance of hepatocellular carcinoma cells via activating AKT phosphorylation by down-regulating miR-34a. Biosci. Rep. 2020, 40, BSR20201627.
- Xu, G.; Zhu, Y.; Liu, H.; Liu, Y.; Zhang, X. LncRNA MIR194-2HG Promotes Cell Proliferation and Metastasis via Regulation of miR-1207-5p/TCF19/Wnt/β-Catenin Signaling in Liver Cancer. Onco. Targets Ther. 2020, 13, 9887–9899.
- Zhu, L.; Yang, N.; Li, C.; Liu, G.; Pan, W.; Li, X. Long noncoding RNA NEAT1 promotes cell proliferation, migration, and invasion in hepatocellular carcinoma through interacting with miR-384. J. Cell. Biochem. 2018, 120, 1997–2006.
- Huang, H.; Chen, J.; Ding, C.M.; Jin, X.; Jia, Z.M.; Peng, J. LncRNA NR2F1-AS1 regulates hepatocellular carcinoma oxaliplatin resistance by targeting ABCC1 via miR-363. J. Cell. Mol. Med. 2018, 22, 3238–3245.
- Qin, M.; Meng, Y.; Luo, C.; He, S.; Qin, F.; Yin, Y.; Huang, J.; Zhao, H.; Hu, J.; Deng, Z.; et al. lncRNA PRR34-AS1 promotes HCC development via modulating Wnt/β-catenin pathway by absorbing miR-296-5p and upregulating E2F2 and SOX12. Mol. Ther. Nucleic Acids 2021, 25, 37–52.
- Sun, L.; Wang, L.; Chen, T.; Shi, Y.; Yao, B.; Liu, Z.; Wang, Y.; Li, Q.; Liu, R.; Niu, Y.; et al. LncRNA RUNX1-IT1 which is downregulated by hypoxia-driven histone deacetylase 3 represses proliferation and cancer stem-like properties in hepatocellular carcinoma cells. Cell Death Dis. 2020, 11, 95.
- Li, Y.; Guo, D.; Zhao, Y.; Ren, M.; Lu, G.; Wang, Y.; Zhang, J.; Mi, C.; He, S.; Lu, X. Long non-coding RNA SNHG5 promotes human hepatocellular carcinoma progression by regulating miR-26a-5p/GSK3β signal pathway. Cell Death Dis. 2018, 9, 888.
- Wu, S.; Chen, S.; Lin, N.; Yang, J. Long non-coding RNA SUMO1P3 promotes hepatocellular carcinoma progression through activating Wnt/β-catenin signalling pathway by targeting miR-320a. J. Cell. Mol. Med. 2020, 24, 3108–3116.
- Zhang, W.; Wu, Y.; Hou, B.; Wang, Y.; Deng, D.; Fu, Z.; Xu, Z. A SOX9-AS1/miR-5590-3p/SOX9 positive feedback loop drives tumor growth and metastasis in hepatocellular carcinoma through the Wnt/β-catenin pathway. Mol. Oncol. 2019, 13, 2194–2210.
- Aggarwal, V.; Tuli, H.S.; Varol, A.; Thakral, F.; Yerer, M.B.; Sak, K.; Varol, M.; Jain, A.; Khan, M.A.; Sethi, G. Role of Reactive Oxygen Species in Cancer Progression: Molecular Mechanisms and Recent Advancements. Biomolecules 2019, 9, 735.
- Li, B.; Cao, Y.; Meng, G.; Qian, L.; Xu, T.; Yan, C.; Luo, O.; Wang, S.; Wei, J.; Ding, Y.; et al. Targeting glutaminase 1 attenuates stemness properties in hepatocellular carcinoma by increasing reactive oxygen species and suppressing Wnt/beta-catenin pathway. EBioMedicine 2019, 39, 239–254.
- Zhu, L.; Yang, X.; Feng, J.; Mao, J.; Zhang, Q.; He, M.; Mi, Y.; Mei, Y.; Jin, G.; Zhang, H. CYP2E1 plays a suppressive role in hepatocellular carcinoma by regulating Wnt/Dvl2/β-catenin signaling. J. Transl. Med. 2022, 20, 194.
- Xu, W.; Zhou, W.; Cheng, M.; Wang, J.; Liu, Z.; He, S.; Luo, X.; Huang, W.; Chen, T.; Yan, W.; et al. Hypoxia activates Wnt/β-catenin signaling by regulating the expression of BCL9 in human hepatocellular carcinoma. Sci. Rep. 2017, 7, 40446.
- To, J.C.; Chiu, A.P.; Tschida, B.R.; Lo, L.H.; Chiu, C.H.; Li, X.X.; Kuka, T.P.; Linden, M.A.; Amin, K.; Chan, W.C.; et al. ZBTB20 regulates WNT/CTNNB1 signalling pathway by suppressing PPARG during hepatocellular carcinoma tumourigenesis. JHEP Rep. 2021, 3, 100223.
- Liu, R.; Li, Y.; Tian, L.; Shi, H.; Wang, J.; Liang, Y.; Sun, B.; Wang, S.; Zhou, M.; Wu, L.; et al. Gankyrin drives metabolic reprogramming to promote tumorigenesis, metastasis and drug resistance through activating β-catenin/c-Myc signaling in human hepatocellular carcinoma. Cancer Lett. 2019, 443, 34–46.
- Delgado, E.; Bahal, R.; Yang, J.; Lee, J.M.; Ly, D.H.; Monga, S.P. β-Catenin knockdown in liver tumor cells by a cell permeable gamma guanidine-based peptide nucleic acid. Curr. Cancer Drug Targets 2013, 13, 867–878.
- Kaur, P.; Mani, S.; Cros, M.P.; Scoazec, J.Y.; Chemin, I.; Hainaut, P.; Herceg, Z. Epigenetic silencing of sFRP1 activates the canonical Wnt pathway and contributes to increased cell growth and proliferation in hepatocellular carcinoma. Tumour Biol. 2012, 33, 325–336.
- Patil, M.A.; Lee, S.A.; Macias, E.; Lam, E.T.; Xu, C.; Jones, K.D.; Ho, C.; Rodriguez-Puebla, M.; Chen, X. Role of cyclin D1 as a mediator of c-Met- and beta-catenin-induced hepatocarcinogenesis. Cancer Res. 2009, 69, 253–261.
- Tang, B.; Tang, F.; Wang, Z.; Qi, G.; Liang, X.; Li, B.; Yuan, S.; Liu, J.; Yu, S.; He, S. Overexpression of CTNND1 in hepatocellular carcinoma promotes carcinous characters through activation of Wnt/β-catenin signaling. J. Exp. Clin. Cancer Res. 2016, 35, 82.
- Qu, B.; Liu, B.R.; Du, Y.J.; Chen, J.; Cheng, Y.Q.; Xu, W.; Wang, X.H. Wnt/β-catenin signaling pathway may regulate the expression of angiogenic growth factors in hepatocellular carcinoma. Oncol. Lett. 2014, 7, 1175–1178.
- Wang, C.; Zhang, R.; Wang, X.; Zheng, Y.; Jia, H.; Li, H.; Wang, J.; Wang, N.; Xiang, F.; Li, Y. Silencing of KIF3B Suppresses Breast Cancer Progression by Regulating EMT and Wnt/β-Catenin Signaling. Front. Oncol. 2020, 10, 597464.
- Ma, B.; Hottiger, M.O. Crosstalk between Wnt/β-Catenin and NF-κB Signaling Pathway during Inflammation. Front. Immunol. 2016, 7, 378.
- Chang, Y.S.; Chou, Y.P.; Chung, C.C.; Lee, Y.T.; Yen, J.C.; Jeng, L.B.; Chang, J.G. Molecular Classification of Hepatocellular Carcinoma Using Wnt–Hippo Signaling Pathway-Related Genes. Cancers 2022, 14, 4580.
- Song, H.; Mak, K.K.; Topol, L.; Yun, K.; Hu, J.; Garrett, L.; Chen, Y.; Park, O.; Chang, J.; Simpson, R.M.; et al. Mammalian Mst1 and Mst2 kinases play essential roles in organ size control and tumor suppression. Proc. Natl. Acad. Sci. USA 2010, 107, 1431–1436.
More
Information
Subjects:
Gastroenterology & Hepatology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
825
Revisions:
2 times
(View History)
Update Date:
17 Jan 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No