Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Gastroenterology & Hepatology
Hepatocellular carcinoma (HCC) is a major cause of cancer death worldwide due to its high rates of tumor recurrence and metastasis. Aberrant Wnt/β-catenin signaling has been shown to play a significant role in HCC development, progression and clinical impact on tumor behavior.
- cancer metabolism
- drug resistance
- metabolic reprogramming
- hepatocellular carcinoma
- Wnt/β-catenin
1. Mutation and Expression Status of the Wnt/β-Catenin Pathway and Its Clinical Significance
Wnt/β-catenin signaling is crucial in contributing to HCC pathogenesis, where genetic mutations and epigenetic alterations are primarily revealed [10]. Activation of the Wnt/β-catenin signaling pathway was discovered in 20–35% of HCC cases, among which most are resulted by gene mutations of the key genes, including CTNNB1, AXIN, and APC [16,17,18]. In this case, CTNNB1 is the gene that specifically encodes β-catenin. Mutation of β-catenin is positively related to HCC progression due to its oncogenic role [19]. To date, mutations at the serine/threonine sites of exon 3 of the β-catenin gene are mostly found to be involved in the phosphorylation and ubiquitination of β-catenin, thus enhancing its nuclear translocation in approximately 20% of HCC cases [20,21]. In addition, conventional and missense mutations have also been reported in other codons of β-catenin [22]. Previous reports showed that conventional mutations at codons 33, 37, 41, and 45 are discovered in over 12% of HCC patients, where missense mutations are observed at codons 32, 34, and 35 [22], which indicates the capability of mutated β-catenin proteins to evade degradation and enter the nucleus [20,23]. It is also noted that tumor cells with aberrant Wnt/β-catenin activation due to the mutation of β-catenin that tend to grow and spread more quickly in HCC [19].
Apart from β-catenin, deregulation of the Wnt/β-catenin signaling pathway is also caused by mutations in protein degradation complexes [24]. These mutations cause dysfunction of the destruction complex and accumulation of β-catenin in the nucleus in approximately 40–70% of HCC cases [10]. One example is the amino acid substitution in armadillo repeats domain 5/6 of β-catenin in human HCC cases [25]. This results in a reduction of APC binding to the degradation complex, which activates the Wnt/β-catenin signaling pathway and enhances targeted gene transcription [25]. It has been reported that a small amount of β-catenin accumulated in the nucleus is sufficient to activate Wnt target genes, suggesting the crucial role of β-catenin in HCC progression [26,27]. However, several studies have shown that the mutation of β-catenin alone is insufficient for promoting HCC in mice, which is different in comparison with humans [27,28], as the tumorigenic potential could be augmented when combined with other oncogenic pathways, such as H-RAS, MET, AKT, or chemicals such as diethylnitrosamine (DEN) [10,29].
In addition, high levels of E-cadherin have been reported to be correlated with the accumulation of β-catenin in both the cytosol and nucleus, that drives the transcription of Wnt target genes [9]. C-Myc and cyclin D, as key Wnt-target genes, not only perform their roles as proto-oncogenes for tumor formation but also regulate liver cancer stem cell (CSC) properties by mediating various signaling pathways involved in cellular differentiation and survival [19,20]. As previously mentioned, HBV and HCV are the causes of HCC, in which they lead to genetic mutations in genes involved in Wnt/β-catenin signaling [9,30]. It is common to find CTNNB1 mutations in HCV-related HCC rather than HBV-related HCC or nonviral HCC [24]. However, mutation of Axin1 is more often found in HBV-related HCC tumors [24]. Interestingly, apart from the mutations of the canonical pathway, Zucman-Rossi et al. suggested that Axin1 mutation also plays a role in exerting oncogenic effects manifested by overexpressing glutamine synthase (GS), leading to β-catenin activation that correlates to the non-canonical pathway [31].
2. Regulation of Wnt/β-Catenin Pathway in HCC
2.1. Epigenetic Regulation of Wnt/β-Catenin
Several epigenetic dysregulations contribute to Wnt/β-catenin activation in HCC. DNA methylation is crucial in maintaining CSC properties, in which its inhibition can influence the fate of cells and gene expressions [32]. For instance, DNA methyltransferase (DNMT) plays a role in catalyzing the transition between a methyl group and DNA, mediating BEX1 expression in HCC [33]. A decrease in DNMT1 results in BEX1 hypomethylation that further enhances the transcription of β-catenin, which causes the activation of the Wnt/β-catenin signaling pathway [33]. Moreover, secreted frizzled-related proteins (SFRPs) negatively regulate Wnt/β-catenin signaling via DNA methylation, representing a leading cause of activating β-catenin activity in HCC [34]. Another study also consistently showed that downregulation of the SFRP family is correlated with Wnt/β-catenin signaling activation, in which SFRP1 and SFRP5 are also found to enhance the progression of HCC [35,36]. Similarly, downregulating Wnt inhibitory factor 1 (WIF1) or Dickkopf-related protein 3 (DKK3) has been proven to result in common consequences for SFRPs [37]. In addition, SOX17 is reported to take part in the aberrant activation of Wnt/β-catenin signaling due to promoter methylation [38]. Silencing of SOX17 could enhance Wnt activity due to the failure in interacting with TCF/LEF, which hinders Wnt-target gene transcription [39]. Apart from the genomic instability caused by DNA hypomethylation, another study showed the involvement of potassium channels in epigenetic regulation of the Wnt/β-catenin pathway [40]. Fan et al. revealed that a decrease in KCNQ1 (potassium voltage-gated channel subfamily Q member 1) causes an increment in Wnt/β-catenin activity via DNA hypermethylation [40].
Furthermore, several alterations through histone modification have been reported in HCC. Enhancer of zeste homologous 2 (EZH2) is a histone methyltransferase that plays a role in catalyzing methylation of histone H3 to achieve repression of Wnt antagonists, promoting Wnt/β-catenin signaling and hepatocarcinogenesis [41,42]. Histone deacetylases (HDAC) have been revealed to interact with EZH2 through its enzymatic role [43]. Specifically, for HDAC8, its upregulation due to the chromatin modifications is coexpressed with the lipogenic transcription factor SREBP1 in HCC mouse models, causing cell cycle arrest and β-catenin activation, which drives NAFLD-induced hepatocarcinogenesis [43]. Moreover, HDAC8 can also bind to pyruvate kinase M2 (PKM2) and subsequently deacetylate the residue K62, prompting the nuclear translocation of PKM2 and the binding of β-catenin that results in Wnt target gene transcription [44]. Similarly, EZH2 overexpression elevated the levels of the oncogene H3K27me3, which silenced Wnt inhibitors, leading to induced cell proliferation with activated β-catenin activity [41].
2.2. Non-Coding RNAs in Regulation of Wnt/β-Catenin
It has been suggested that microRNAs (miRNAs) and long noncoding RNAs (lncRNAs) are critical regulators associated with various tumors, in which they are negatively regulated [45]. Dysregulation of miRNAs and lncRNAs could lead to tumorigenesis in HCC. LncRNA-miRNA binding yields a complete endogenous RNA (ceRNA) that can avoid messenger RNA (mRNA) recognition and further silencing effects, known as the “sponge effect” [45]. Mounting evidence suggests that miRNA sponges are involved in Wnt/β-catenin signaling and are associated with HCC progression (Table 1). For example, LINC00355 and LINC01278 are negative regulators of miR-217-5p and miR-1258, respectively [46,47]. Overexpression of lncRNAs downregulate the corresponding miRNAs and further activates Wnt/β-catenin signaling, resulting in increased levels of Wnt target gene transcription and metastatic ability of HCC cells [46,47]. Additionally, upregulation of LINC00662 in HCC induced WNT3A secretion with miR-15a/16/107 binding, resulting in the activation of Wnt/β-catenin and polarizes M2 macrophage [48]. Similarly, overexpressing FEZF1-AS1 negatively regulates the level of miR-107, which inhibits the activation of Wnt/β-catenin signaling, while downregulation of FEZF1-AS1 enhances the expression of β-catenin [49]. Furthermore, both miR-122 and miR-148a were found to contribute to liver cancers by binding to the 3′-untranslated region (3′-UTR) site of Wnt1, suppressing the level of β-catenin and inhibiting Wnt-target gene transcription [50,51]. Furthermore, a decrease in these miRNA levels could cause excess Wnt/β-catenin signaling and increase EMT [50]. All the above mentioned enhance the progression of HCC. As a tumor suppressor, miR-34a is reported in mice and HCC patients and found to be upregulated through activated Wnt/β-catenin signaling [52]. In addition, overexpression of miR-145 has been shown to diminish the level of β-catenin, suppressing HCC cell growth [53]. To sum up, Wnt/β-catenin signaling is tightly regulated by DNA methylation, histone modification and non-coding RNAs in HCC (Figure 1).
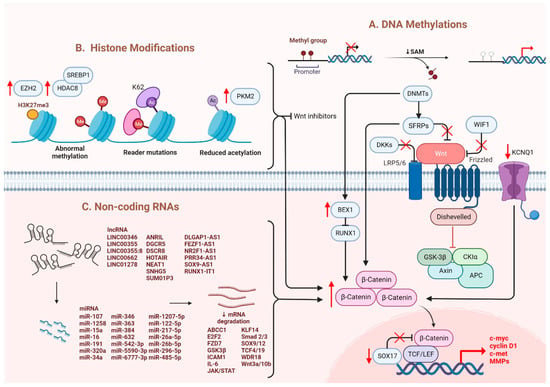
Figure 1. Regulation of Wnt/β-catenin signaling in HCC. Wnt/β-catenin signaling in HCC is regulated by (A) DNA methylation, (B) histone modification and (C) non-coding RNAs.
Table 1. The list of lncRNAs and their related miRNAs in regulation of Wnt/β-catenin signaling in HCC.
| LncRNA | miRNA | Targets | Ref. |
|---|---|---|---|
| LINC00346 | miR-542-3p | FZD7, WDR18 | [54] |
| LINC00355 | miR-217-5p | GSK3β, c-myc, CCND1 | [46] |
| LINC00355:8 | miR-6777-3p | Wnt10b | [55] |
| LINC00662 | miR-15a, miR-16, miR-107 | Wnt3a | [48] |
| LINC01278 | miR-1258 | TCF-4, Smad2/3 | [47] |
| ANRIL | miR-191, miR-122-5p |
CCND1, p53, p21, MMP-2, MMP-9, Vimentin | [56,57] |
| DGCR5 | miR-346 | KLF14 | [58] |
| DSCR8 | miR-485-5p | FZD7 | [59] |
| DLGAP1-AS1 | miR-26a-5p, miR-26b-5p |
IL-6, JAK2, STAT3 | [60] |
| FEZF1-AS1 | miR-107 | Wnt3a, ICAM1, Vimentin | [49] |
| HOTAIR | miR-34a | Akt | [61] |
| MIR194-2HG | miR-1207-5p | TCF19 | [62] |
| NEAT1 | miR-384 | Wnt | [63] |
| NR2F1-AS1 | miR-363 | ABCC1 | [64] |
| PRR34-AS1 | miR-296-5p | E2F2, SOX12 | [65] |
| RUNX1-IT1 | miR-632 | GSK3β | [66] |
| SNHG5 | miR-26a-5p | GSK3β | [67] |
| SUMO1P3 | miR-320a | C-myc, CCND1 | [68] |
| SOX9-AS1 | miR-5590-3p | SOX9 | [69] |
2.3. Other Molecules Involved in the Regulation of Wnt/β-Catenin
Apart from genetic mutations and epigenetic dysregulation, other molecules/pathways were identified to regulate Wnt/β-catenin signaling. In normoric environment, ROS is maintained at a low level; whereas a steady increase of ROS level promotes cancer development and progression [70]. A recent study showed that Wnt/β-catenin signaling was suppressed upon elevation of intracellular ROS level [19,71]. In HCC, glutaminase 1 (GLS1) is upregulated which augmented liver CSC properties with increased expression of CSC markers via suppression of ROS level [19,71]. Likewise, another study also showed that ROS accumulation due to the overexpression of Cytochrome P450 2E1 (CYP2E1) decreased the activity of Wnt/β-catenin signaling through the degradation of DVL2 in HCC [72]. Hypoxia also plays a crucial role in the activation of Wnt/β-catenin signaling. Hypoxia-inducible factor 1-alpha (HIF1α), a hypoxia-inducible factor, regulates transcription in hypoxic environments and is also reported to mediate the expression of B-cell lymphoma 9 (BCL9) [9,73]. BCL9 can coactivate with HIF1α to enhance the transcriptional activity of β-catenin regardless of whether genetic mutations occur, resulting in activation of Wnt/β-catenin signaling and leading to HCC progression [73]. Furthermore, ZBTB20 has been reported in liver tumorigenesis with its role in suppressing PPARG expression and inhibiting proteasomal degradation of the β-catenin destruction complex [74]. Overall, once the nuclear translocation of β-catenin is achieved, the expression levels of the downstream genes involved in EMT are modulated and enhanced, causing hepatocarcinogenesis [17]. C-Myc is the most critical gene induced by activated Wnt/β-catenin signaling, which enhances the mechanisms of glycolysis and glutaminolysis [75]. This is followed by cyclin D1, which has been reported to be enhanced in both mouse and human HCC [76,77]. Specifically, overexpression of c-Met and cyclin D1 triggers the development of liver tumors and decreases survival in mice [78]. It is also noted that upregulation of cyclin D1 enhances tumor metastatic ability [79]. Additionally, studies have discovered that GS and VEGF are also involved in modulating the downstream effects of activated Wnt and assisting in angiogenesis [80], as the upregulation of multiple matrix metalloproteinases (MMPs), including MMP2 and MMP9, is associated with tumor metastasis [81]. Apart from gene regulation, aberrant β-catenin signaling also negatively regulates certain signaling cascades: for example, the suppression of NF-κB cascade in the liver [82]. Moreover, the crosstalk between Wnt and Hippo signaling pathways has been observed in HCC. Recent study showed that Wnt-Hippo signature related genes may be a potential markers for prediction of immune infiltration in HCC [83]. Notably, aberrant activation of β-catenin caused by the deletion of mammalian STE20-like protein kinase 1/2 (Mst1/2) promotes tumor growth, indicating the co-expression of YAP and β-catenin in HCC [84].
This entry is adapted from the peer-reviewed paper 10.3390/cancers14215468
This entry is offline, you can click here to edit this entry!