 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Sofia Vrettou | -- | 4751 | 2022-12-24 13:55:53 | | | |
| 2 | Amina Yu | -10 word(s) | 4741 | 2022-12-26 02:12:34 | | | | |
| 3 | Amina Yu | Meta information modification | 4741 | 2022-12-26 02:14:18 | | |
Video Upload Options
Redox post-translational modifications are derived from fluctuations in the redox potential and modulate protein function, localization, activity and structure. Amongst the oxidative reversible modifications, the S-glutathionylation of proteins was the first to be characterized as a post-translational modification, which primarily protects proteins from irreversible oxidation. S-nitrosylation, another post-translational modification, was identified in the past, but it was re-introduced as a prototype cell-signaling mechanism, one that tightly regulates core processes within the cell’s sub-compartments, especially in mitochondria. S-glutathionylation and S-nitrosylation are modulated by fluctuations in reactive oxygen and nitrogen species and, in turn, orchestrate mitochondrial bioenergetics machinery, morphology, nutrients metabolism and apoptosis.
1. Introduction
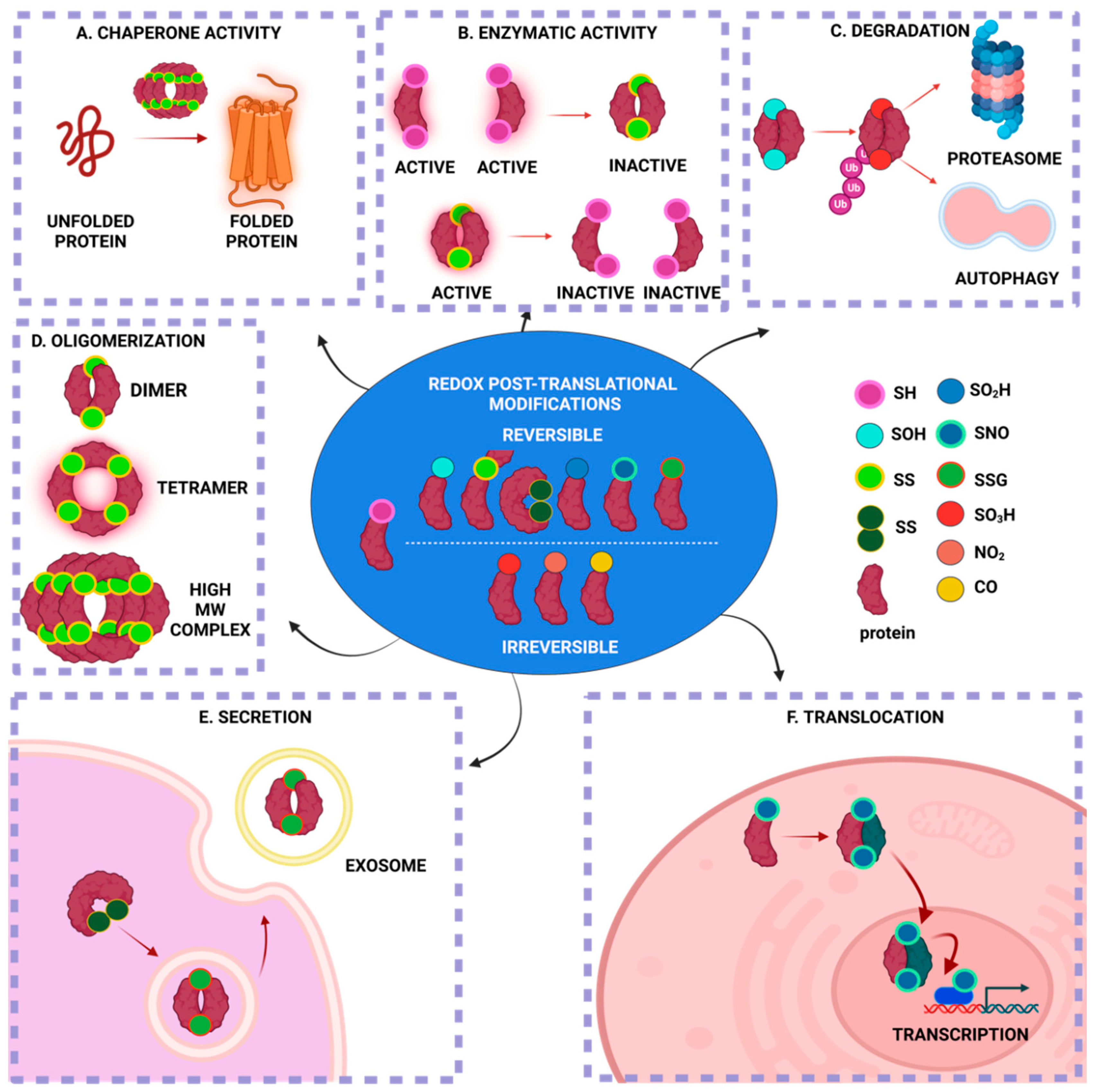
2. Protein S-Glutathionylation

2.1. Regulation of OXPHOS System by S-Glutathionylation
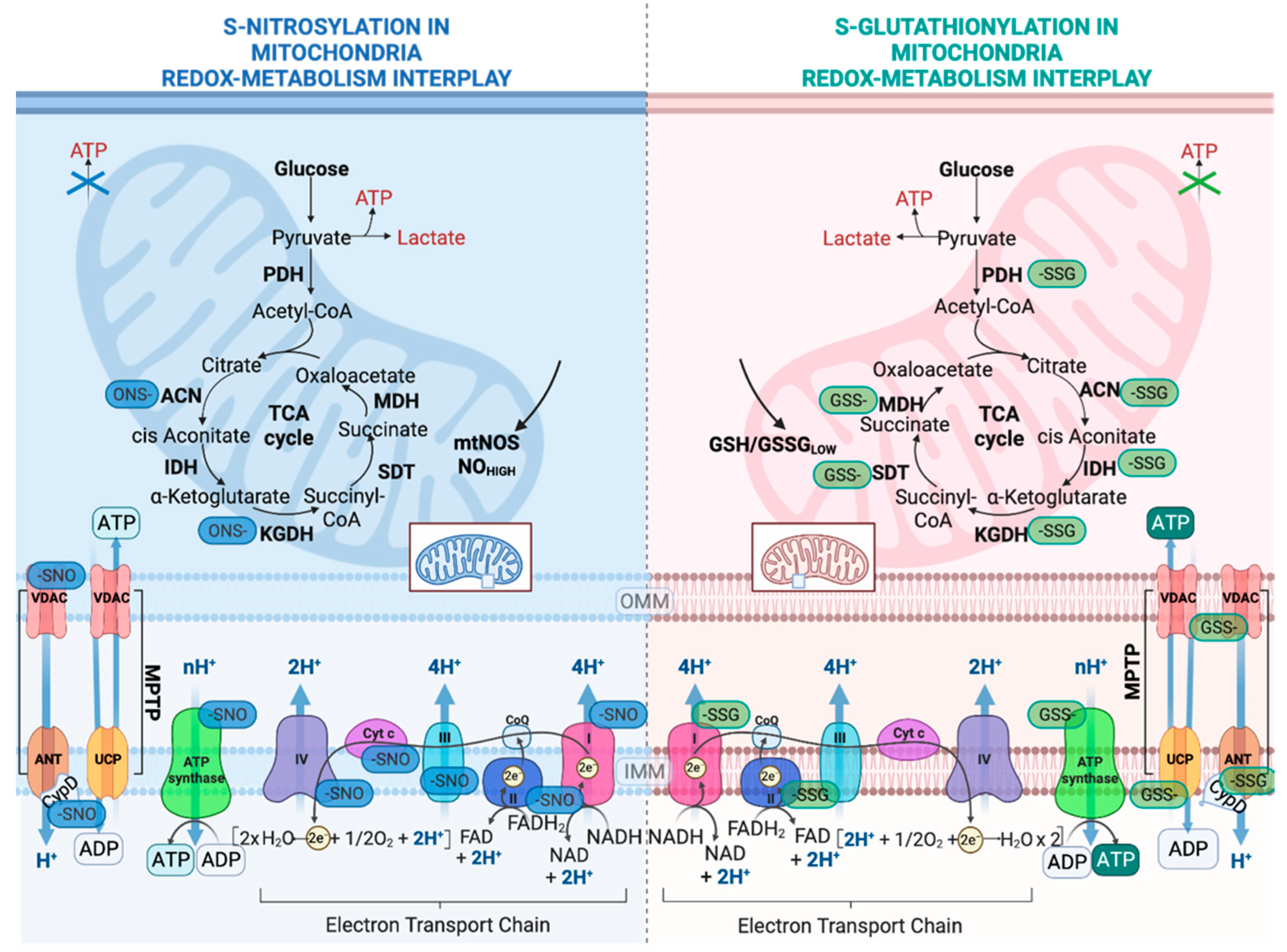
2.2. Regulation of Nutrient Metabolism by S-Glutathionylation
2.3. Regulation of Mitochondrial Permeability Transition Pore and Apoptosis by S-Glutathionylation
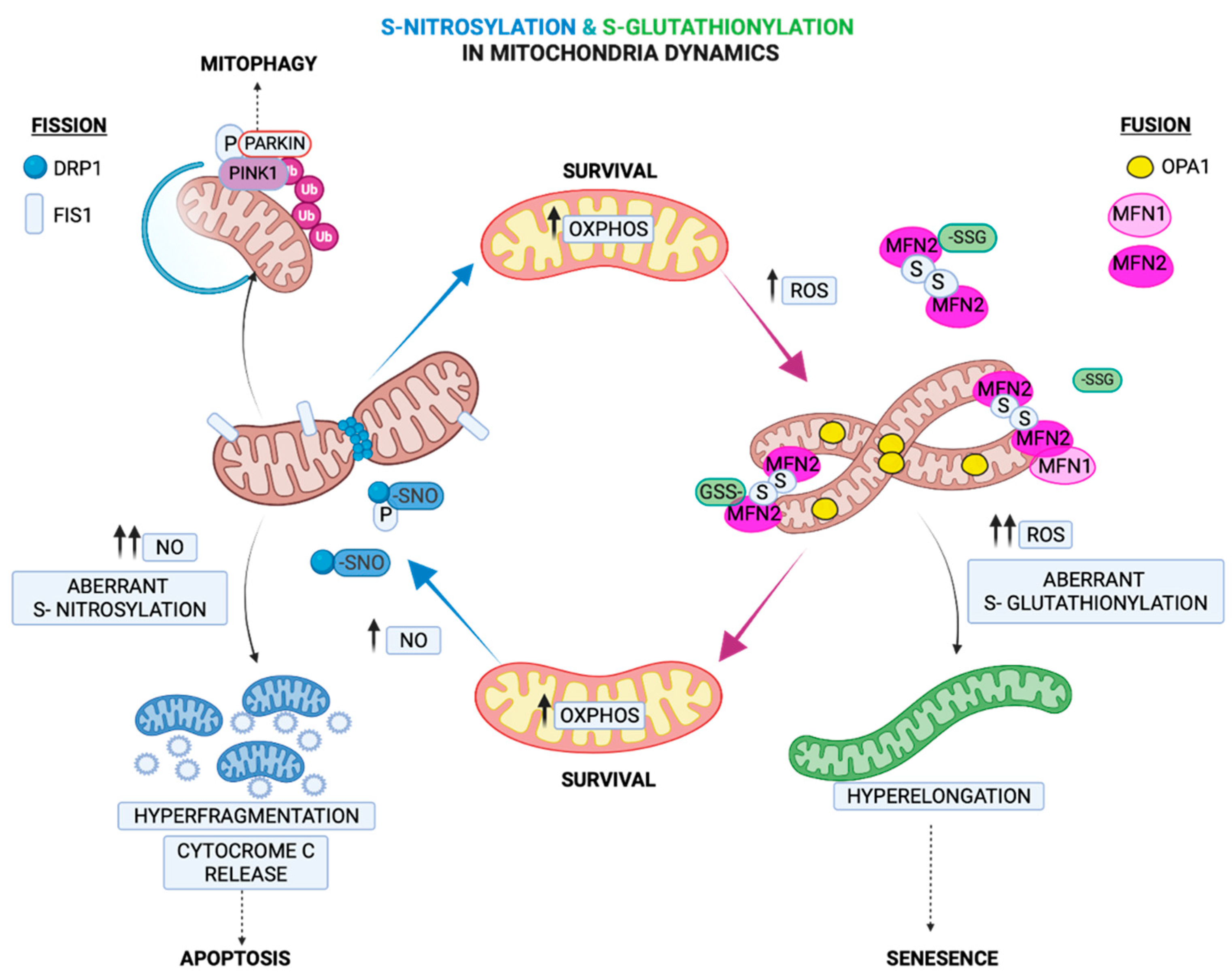
2.4. Regulation of Mitochondria Morphology by S-Glutathionylation
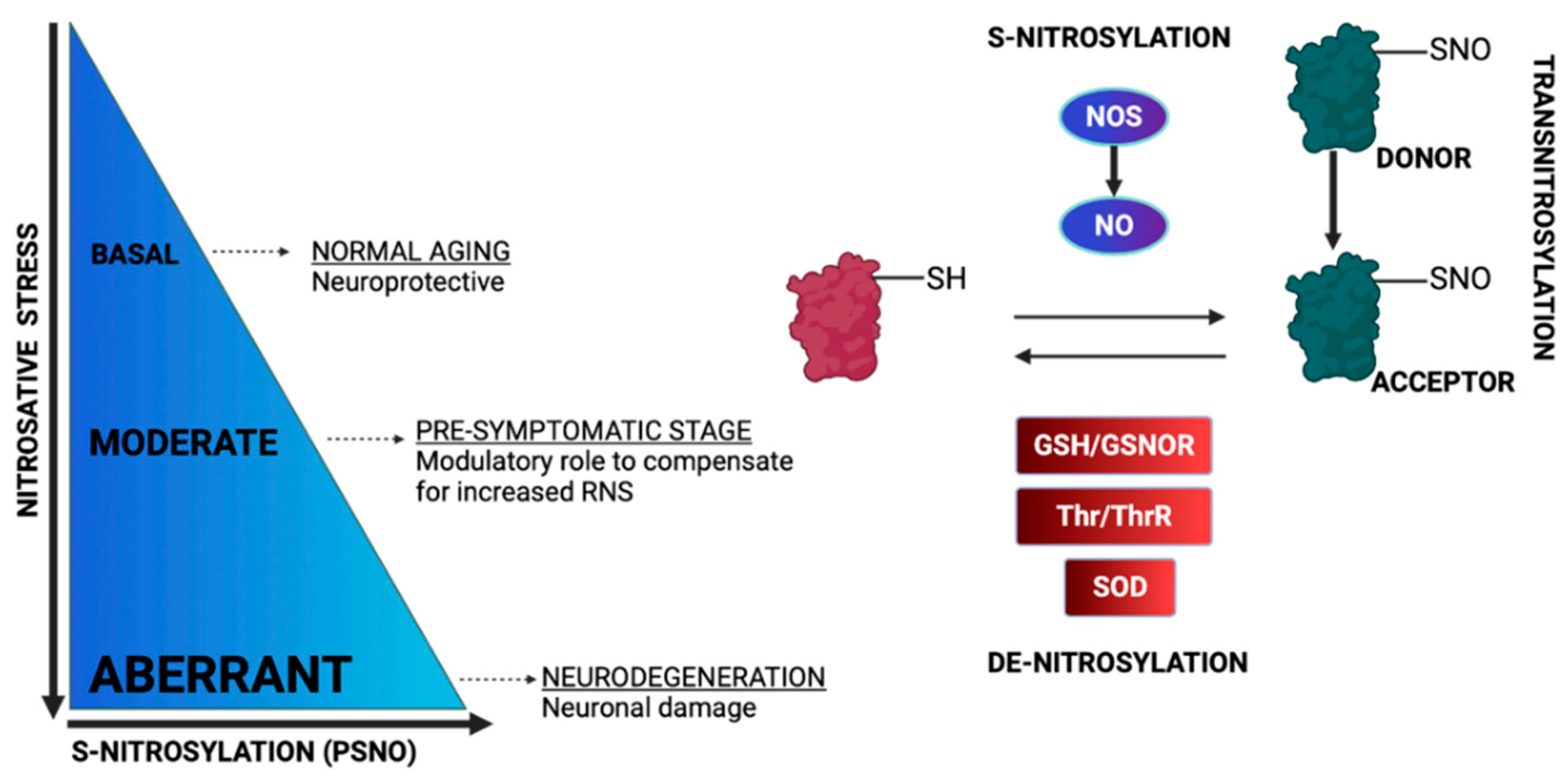
3. Protein S-Nitrosylation
3.1. Regulation of the OXPHOS System by S-Nitrosylation
3.2. Regulation of Metabolic Enzymes by S-Nitrosylation
3.3. Regulation of Apoptosis by S-Nitrosylation
-
Mitochondrial permeability transition pores (MPTPs) are formed under oxidative stress and modulate the redox potential. Under oxidative stress, MPTP increases the permeability relative to high molecular weight macromolecules, leading to mitochondria swelling and necroptosis [100]. Under the basal levels of NO, cyclophilin D (CYPD), critical for an MPTP opening, can be S-nitrosylated [101]. This inhibits its interaction with MPTPs, preventing pore opening and protecting cells during stress. Conversely, under the increased production of NO, peroxynitrite is being produced by excessive ROS, which leads to the opening of MPTPs, the ablation of ATP production and necrosis [102].
-
VDAC has multiple roles. Its localization at the mitochondrial outer membrane allows VDAC to regulate the fluxes of metabolites. At the same time, VDAC modulates cytochrome c, which activates caspase-dependent apoptotic cell death [103]. Similarly to MPTP regulation by NO, VDAC S-nitrosylation at basal levels of NO inhibits its function, protecting cells from apoptosis, whereas elevated levels of NO upregulate VDAC functions [104].
-
Crucial to their role in apoptosis, caspase-3 and caspase-9 are S-nitrosylated in the absence of apoptotic triggers, while their denitrosylation leads to their activation upon the activation of the Fas receptor [105].
-
Another component of the outer mitochondrial membrane with an anti-apoptotic role involves BCL2 family proteins, which interfere with apoptosis by regulating the release of cytochrome c [106]. Upon apoptotic signaling, the S-nitrosylation of BCL-2 inhibits its ubiquitination and degradation, allowing it to exert anti-apoptotic protection [107].
3.4. Regulation of Fission by S-Nitrosylation
References
- Weidinger, A.; Kozlov, A.V. Biological Activities of Reactive Oxygen and Nitrogen Species: Oxidative Stress versus Signal Transduction. Biomolecules 2015, 5, 472–484.
- Santos, A.L.; Lindner, A.B. Protein Posttranslational Modifications: Roles in Aging and Age-Related Disease. Oxidative Med. Cell Longev. 2017, 2017, 5716409.
- Beckhauser, T.F.; Francis-Oliveira, J.; De Pasquale, R. Reactive Oxygen Species: Physiological and Physiopathological Effects on Synaptic Plasticity. J. Exp. Neurosci. 2016, 10, 23–48.
- Jones, D.P. Redefining oxidative stress. Antioxid. Redox. Signal. 2006, 8, 1865–1879.
- Ray, P.D.; Huang, B.W.; Tsuji, Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell Signal. 2012, 24, 981–990.
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13.
- Lambeth, J.D.; Neish, A.S. Nox enzymes and new thinking on reactive oxygen: A double-edged sword revisited. Annu. Rev. Pathol. 2014, 9, 119–145.
- Ma, M.W.; Wang, J.; Zhang, Q.; Wang, R.; Dhandapani, K.M.; Vadlamudi, R.K.; Brann, D.W. NADPH oxidase in brain injury and neurodegenerative disorders. Mol. Neurodegener. 2017, 12, 7.
- Di Meo, S.; Reed, T.T.; Venditti, P.; Victor, V.M. Role of ROS and RNS Sources in Physiological and Pathological Conditions. Oxid. Med. Cell Longev. 2016, 2016, 1245049.
- Birben, E.; Sahiner, U.M.; Sackesen, C.; Erzurum, S.; Kalayci, O. Oxidative stress and antioxidant defense. World Allergy Organ. J. 2012, 5, 9–19.
- Moldogazieva, N.T.; Mokhosoev, I.M.; Feldman, N.B.; Lutsenko, S.V. ROS and RNS signalling: Adaptive redox switches through oxidative/nitrosative protein modifications. Free Radic. Res. 2018, 52, 507–543.
- Paul, B.D.; Sbodio, J.I.; Snyder, S.H. Cysteine Metabolism in Neuronal Redox Homeostasis. Trends. Pharm. Sci. 2018, 39, 513–524.
- Sbodio, J.I.; Snyder, S.H.; Paul, B.D. Redox Mechanisms in Neurodegeneration: From Disease Outcomes to Therapeutic Opportunities. Antioxid. Redox. Signal. 2019, 30, 1450–1499.
- Stadtman, E.R.; Levine, R.L. Free radical-mediated oxidation of free amino acids and amino acid residues in proteins. Amino. Acids. 2003, 25, 207–218.
- Davies, M.J. Protein oxidation and peroxidation. Biochem. J. 2016, 473, 805–825.
- Wall, S.B.; Oh, J.Y.; Diers, A.R.; Landar, A. Oxidative modification of proteins: An emerging mechanism of cell signaling. Front. Physiol. 2012, 3, 369.
- Wani, R.; Nagata, A.; Murray, B.W. Protein redox chemistry: Post-translational cysteine modifications that regulate signal transduction and drug pharmacology. Front. Pharmacol. 2014, 5, 224.
- Halloran, M.; Parakh, S.; Atkin, J.D. The role of s-nitrosylation and s-glutathionylation of protein disulphide isomerase in protein misfolding and neurodegeneration. Int. J. Cell Biol. 2013, 2013, 797914.
- Chung, H.S.; Wang, S.B.; Venkatraman, V.; Murray, C.I.; Van Eyk, J.E. Cysteine oxidative posttranslational modifications: Emerging regulation in the cardiovascular system. Circ. Res. 2013, 112, 382–392.
- Fra, A.; Yoboue, E.D.; Sitia, R. Cysteines as Redox Molecular Switches and Targets of Disease. Front. Mol. Neurosci. 2017, 10, 167.
- Davies, M.J. The oxidative environment and protein damage. Biochim. Biophys. Acta 2005, 1703, 93–109.
- Pajares, M.; Jimenez-Moreno, N.; Dias, I.H.K.; Debelec, B.; Vucetic, M.; Fladmark, K.E.; Basaga, H.; Ribaric, S.; Milisav, I.; Cuadrado, A. Redox control of protein degradation. Redox. Biol. 2015, 6, 409–420.
- Song, H.; Liu, B.; Huai, W.; Yu, Z.; Wang, W.; Zhao, J.; Han, L.; Jiang, G.; Zhang, L.; Gao, C.; et al. The E3 ubiquitin ligase TRIM31 attenuates NLRP3 inflammasome activation by promoting proteasomal degradation of NLRP3. Nat. Commun. 2016, 7, 13727.
- Muronetz, V.I.; Medvedeva, M.V.; Sevostyanova, I.A.; Schmalhausen, E.V. Modification of Glyceraldehyde-3-Phosphate Dehydrogenase with Nitric Oxide: Role in Signal Transduction and Development of Apoptosis. Biomolecules 2021, 11, 1656.
- Cha, S.J.; Kim, H.; Choi, H.J.; Lee, S.; Kim, K. Protein Glutathionylation in the Pathogenesis of Neurodegenerative Diseases. Oxid. Med. Cell Longev. 2017, 2017, 2818565.
- Montagna, C.; Cirotti, C.; Rizza, S.; Filomeni, G. When S-Nitrosylation Gets to Mitochondria: From Signaling to Age-Related Diseases. Antioxid. Redox. Signal. 2020, 32, 884–905.
- Nakamura, T.; Tu, S.; Akhtar, M.W.; Sunico, C.R.; Okamoto, S.; Lipton, S.A. Aberrant protein s-nitrosylation in neurodegenerative diseases. Neuron 2013, 78, 596–614.
- Mailloux, R.J.; Treberg, J.R. Protein S-glutathionlyation links energy metabolism to redox signaling in mitochondria. Redox. Biol. 2016, 8, 110–118.
- Rizza, S.; Cardaci, S.; Montagna, C.; Di Giacomo, G.; De Zio, D.; Bordi, M.; Maiani, E.; Campello, S.; Borreca, A.; Puca, A.A.; et al. S-nitrosylation drives cell senescence and aging in mammals by controlling mitochondrial dynamics and mitophagy. Proc. Natl. Acad. Sci. USA 2018, 115, E3388–E3397.
- Zahid, S.; Khan, R.; Oellerich, M.; Ahmed, N.; Asif, A.R. Differential S-nitrosylation of proteins in Alzheimer’s disease. Neuroscience 2014, 256, 126–136.
- Sultana, R.; Butterfield, D.A. Oxidative modification of brain proteins in Alzheimer’s disease: Perspective on future studies based on results of redox proteomics studies. J. Alzheimers Dis. 2013, 33 (Suppl. S1), S243–S251.
- Sabens Liedhegner, E.A.; Gao, X.H.; Mieyal, J.J. Mechanisms of altered redox regulation in neurodegenerative diseases--focus on S--glutathionylation. Antioxid. Redox. Signal. 2012, 16, 543–566.
- Nakamura, T.; Lipton, S.A. Emerging role of protein-protein transnitrosylation in cell signaling pathways. Antioxid. Redox. Signal. 2013, 18, 239–249.
- Moosmann, B.; Behl, C. Antioxidants as treatment for neurodegenerative disorders. Expert Opin. Investig. Drugs 2002, 11, 1407–1435.
- Mailloux, R.J.; Jin, X.; Willmore, W.G. Redox regulation of mitochondrial function with emphasis on cysteine oxidation reactions. Redox. Biol. 2014, 2, 123–139.
- Dalle-Donne, I.; Rossi, R.; Giustarini, D.; Colombo, R.; Milzani, A. S-glutathionylation in protein redox regulation. Free Radic. Biol. Med. 2007, 43, 883–898.
- Kang, P.T.; Zhang, L.; Chen, C.L.; Chen, J.; Green, K.B.; Chen, Y.R. Protein thiyl radical mediates S-glutathionylation of complex I. Free Radic. Biol. Med. 2012, 53, 962–973.
- Ziegler, D.M. Role of reversible oxidation-reduction of enzyme thiols-disulfides in metabolic regulation. Annu. Rev. Biochem. 1985, 54, 305–329.
- Gallogly, M.M.; Starke, D.W.; Mieyal, J.J. Mechanistic and kinetic details of catalysis of thiol-disulfide exchange by glutaredoxins and potential mechanisms of regulation. Antioxid. Redox. Signal. 2009, 11, 1059–1081.
- Stroher, E.; Millar, A.H. The biological roles of glutaredoxins. Biochem. J. 2012, 446, 333–348.
- Mitra, S.; Elliott, S.J. Oxidative disassembly of the cluster of human Grx2 and redox regulation in the mitochondria. Biochemistry 2009, 48, 3813–3815.
- Beer, S.M.; Taylor, E.R.; Brown, S.E.; Dahm, C.C.; Costa, N.J.; Runswick, M.J.; Murphy, M.P. Glutaredoxin 2 catalyzes the reversible oxidation and glutathionylation of mitochondrial membrane thiol proteins: Implications for mitochondrial redox regulation and antioxidant DEFENSE. J. Biol. Chem. 2004, 279, 47939–47951.
- Mailloux, R.J.; Xuan, J.Y.; Beauchamp, B.; Jui, L.; Lou, M.; Harper, M.E. Glutaredoxin-2 is required to control proton leak through uncoupling protein-3. J. Biol. Chem. 2013, 288, 8365–8379.
- Klaus, A.; Zorman, S.; Berthier, A.; Polge, C.; Ramirez, S.; Michelland, S.; Seve, M.; Vertommen, D.; Rider, M.; Lentze, N.; et al. Glutathione S-transferases interact with AMP-activated protein kinase: Evidence for S-glutathionylation and activation in vitro. PLoS ONE 2013, 8, e62497.
- Raza, H. Dual localization of glutathione S-transferase in the cytosol and mitochondria: Implications in oxidative stress, toxicity and disease. FEBS J. 2011, 278, 4243–4251.
- Park, J.W.; Mieyal, J.J.; Rhee, S.G.; Chock, P.B. Deglutathionylation of 2-Cys peroxiredoxin is specifically catalyzed by sulfiredoxin. J. Biol. Chem. 2009, 284, 23364–23374.
- Aon, M.A.; Cortassa, S.; Maack, C.; O’Rourke, B. Sequential opening of mitochondrial ion channels as a function of glutathione redox thiol status. J. Biol. Chem. 2007, 282, 21889–21900.
- Hurd, T.R.; Requejo, R.; Filipovska, A.; Brown, S.; Prime, T.A.; Robinson, A.J.; Fearnley, I.M.; Murphy, M.P. Complex I within oxidatively stressed bovine heart mitochondria is glutathionylated on Cys-531 and Cys-704 of the 75-kDa subunit: Potential role of CYS residues in decreasing oxidative damage. J. Biol. Chem. 2008, 283, 24801–24815.
- Chen, C.L.; Zhang, L.; Yeh, A.; Chen, C.A.; Green-Church, K.B.; Zweier, J.L.; Chen, Y.R. Site-specific S-glutathiolation of mitochondrial NADH ubiquinone reductase. Biochemistry 2007, 46, 5754–5765.
- Diotte, N.M.; Xiong, Y.; Gao, J.; Chua, B.H.; Ho, Y.S. Attenuation of doxorubicin-induced cardiac injury by mitochondrial glutaredoxin 2. Biochim. Biophys. Acta 2009, 1793, 427–438.
- Taylor, E.R.; Hurrell, F.; Shannon, R.J.; Lin, T.K.; Hirst, J.; Murphy, M.P. Reversible glutathionylation of complex I increases mitochondrial superoxide formation. J. Biol. Chem. 2003, 278, 19603–19610.
- Tretter, L.; Adam-Vizi, V. Inhibition of Krebs cycle enzymes by hydrogen peroxide: A key role of -ketoglutarate dehydrogenase in limiting NADH production under oxidative stress. J. Neurosci. 2000, 20, 8972–8979.
- Garcia, J.; Han, D.; Sancheti, H.; Yap, L.P.; Kaplowitz, N.; Cadenas, E. Regulation of mitochondrial glutathione redox status and protein glutathionylation by respiratory substrates. J. Biol. Chem. 2010, 285, 39646–39654.
- Korshunov, S.S.; Skulachev, V.P.; Starkov, A.A. High protonic potential actuates a mechanism of production of reactive oxygen species in mitochondria. FEBS Lett. 1997, 416, 15–18.
- Bulteau, A.L.; Lundberg, K.C.; Ikeda-Saito, M.; Isaya, G.; Szweda, L.I. Reversible redox-dependent modulation of mitochondrial aconitase and proteolytic activity during in vivo cardiac ischemia/reperfusion. Proc. Natl. Acad. Sci. USA 2005, 102, 5987–5991.
- Kil, I.S.; Park, J.W. Regulation of mitochondrial NADP+-dependent isocitrate dehydrogenase activity by glutathionylation. J. Biol. Chem. 2005, 280, 10846–10854.
- McLain, A.L.; Szweda, P.A.; Szweda, L.I. alpha-Ketoglutarate dehydrogenase: A mitochondrial redox sensor. Free Radic. Res. 2011, 45, 29–36.
- O’Brien, M.; Chalker, J.; Slade, L.; Gardiner, D.; Mailloux, R.J. Protein S-glutathionylation alters superoxide/hydrogen peroxide emission from pyruvate dehydrogenase complex. Free Radic. Biol. Med. 2017, 106, 302–314.
- Mailloux, R.J.; Xuan, J.Y.; McBride, S.; Maharsy, W.; Thorn, S.; Holterman, C.E.; Kennedy, C.R.; Rippstein, P.; de Kemp, R.; da Silva, J.; et al. Glutaredoxin-2 is required to control oxidative phosphorylation in cardiac muscle by mediating deglutathionylation reactions. J. Biol. Chem. 2014, 289, 14812–14828.
- Muoio, D.M. Metabolic inflexibility: When mitochondrial indecision leads to metabolic gridlock. Cell 2014, 159, 1253–1262.
- Giangregorio, N.; Palmieri, F.; Indiveri, C. Glutathione controls the redox state of the mitochondrial carnitine/acylcarnitine carrier Cys residues by glutathionylation. Biochim. Biophys. Acta 2013, 1830, 5299–5304.
- Zhang, J.; Ye, Z.W.; Singh, S.; Townsend, D.M.; Tew, K.D. An evolving understanding of the S-glutathionylation cycle in pathways of redox regulation. Free Radic. Biol. Med. 2018, 120, 204–216.
- Cadenas, S. Mitochondrial uncoupling, ROS generation and cardioprotection. Biochim. Biophys. Acta Bioenerg. 2018, 1859, 940–950.
- Bouillaud, F.; Alves-Guerra, M.C.; Ricquier, D. UCPs, at the interface between bioenergetics and metabolism. Biochim. Biophys. Acta 2016, 1863, 2443–2456.
- Mailloux, R.J.; Seifert, E.L.; Bouillaud, F.; Aguer, C.; Collins, S.; Harper, M.E. Glutathionylation acts as a control switch for uncoupling proteins UCP2 and UCP3. J. Biol. Chem. 2011, 286, 21865–21875.
- Nunnari, J.; Suomalainen, A. Mitochondria: In sickness and in health. Cell 2012, 148, 1145–1159.
- Kowaltowski, A.J.; Netto, L.E.; Vercesi, A.E. The thiol-specific antioxidant enzyme prevents mitochondrial permeability transition. Evidence for the participation of reactive oxygen species in this mechanism. J. Biol. Chem. 1998, 273, 12766–12769.
- Queiroga, C.S.; Almeida, A.S.; Martel, C.; Brenner, C.; Alves, P.M.; Vieira, H.L. Glutathionylation of adenine nucleotide translocase induced by carbon monoxide prevents mitochondrial membrane permeabilization and apoptosis. J. Biol. Chem. 2010, 285, 17077–17088.
- Skulachev, V.P. Bioenergetic aspects of apoptosis, necrosis and mitoptosis. Apoptosis 2006, 11, 473–485.
- Itani, H.A.; Dikalova, A.E.; McMaster, W.G.; Nazarewicz, R.R.; Bikineyeva, A.T.; Harrison, D.G.; Dikalov, S.I. Mitochondrial Cyclophilin D in Vascular Oxidative Stress and Hypertension. Hypertension 2016, 67, 1218–1227.
- Thiriveedi, V.R.; Mattam, U.; Pattabhi, P.; Bisoyi, V.; Talari, N.K.; Krishnamoorthy, T.; Sepuri, N.B.V. Glutathionylated and Fe-S cluster containing hMIA40 (CHCHD4) regulates ROS and mitochondrial complex III and IV activities of the electron transport chain. Redox. Biol. 2020, 37, 101725.
- Liesa, M.; Shirihai, O.S. Mitochondrial dynamics in the regulation of nutrient utilization and energy expenditure. Cell Metab. 2013, 17, 491–506.
- Willems, P.H.; Rossignol, R.; Dieteren, C.E.; Murphy, M.P.; Koopman, W.J. Redox Homeostasis and Mitochondrial Dynamics. Cell Metab. 2015, 22, 207–218.
- Shutt, T.; Geoffrion, M.; Milne, R.; McBride, H.M. The intracellular redox state is a core determinant of mitochondrial fusion. EMBO Rep. 2012, 13, 909–915.
- Che, L.; Yang, C.L.; Chen, Y.; Wu, Z.L.; Du, Z.B.; Wu, J.S.; Gan, C.L.; Yan, S.P.; Huang, J.; Guo, N.J.; et al. Mitochondrial redox-driven mitofusin 2 S-glutathionylation promotes neuronal necroptosis via disrupting ER-mitochondria crosstalk in cadmium-induced neurotoxicity. Chemosphere 2021, 262, 127878.
- Martinez-Ruiz, A.; Araujo, I.M.; Izquierdo-Alvarez, A.; Hernansanz-Agustin, P.; Lamas, S.; Serrador, J.M. Specificity in S-nitrosylation: A short-range mechanism for NO signaling? Antioxid. Redox. Signal. 2013, 19, 1220–1235.
- Martinez-Ruiz, A.; Lamas, S. Signalling by NO-induced protein S-nitrosylation and S-glutathionylation: Convergences and divergences. Cardiovasc. Res. 2007, 75, 220–228.
- Fernando, V.; Zheng, X.; Walia, Y.; Sharma, V.; Letson, J.; Furuta, S. S-Nitrosylation: An Emerging Paradigm of Redox Signaling. Antioxidants 2019, 8, 404.
- Sun, J.; Steenbergen, C.; Murphy, E. S-nitrosylation: NO-related redox signaling to protect against oxidative stress. Antioxid. Redox. Signal. 2006, 8, 1693–1705.
- Lancaster, J.R., Jr. Nitric oxide: A brief overview of chemical and physical properties relevant to therapeutic applications. Future Sci. OA 2015, 1, FSO59.
- Beltran, B.; Orsi, A.; Clementi, E.; Moncada, S. Oxidative stress and S-nitrosylation of proteins in cells. Br. J. Pharm. 2000, 129, 953–960.
- Whiteman, M.; Chua, Y.L.; Zhang, D.; Duan, W.; Liou, Y.C.; Armstrong, J.S. Nitric oxide protects against mitochondrial permeabilization induced by glutathione depletion: Role of S-nitrosylation? Biochem. Biophys. Res. Commun. 2006, 339, 255–262.
- Stomberski, C.T.; Hess, D.T.; Stamler, J.S. Protein S-Nitrosylation: Determinants of Specificity and Enzymatic Regulation of S-Nitrosothiol-Based Signaling. Antioxid. Redox. Signal. 2019, 30, 1331–1351.
- Zhang, Y.; Wu, K.; Su, W.; Zhang, D.F.; Wang, P.; Qiao, X.; Yao, Q.; Yuan, Z.; Yao, Y.G.; Liu, G.; et al. Increased GSNOR Expression during Aging Impairs Cognitive Function and Decreases S-Nitrosation of CaMKIIalpha. J. Neurosci. 2017, 37, 9741–9758.
- Nakamura, T.; Lipton, S.A. Cell death: Protein misfolding and neurodegenerative diseases. Apoptosis 2009, 14, 455–468.
- Ozawa, K.; Komatsubara, A.T.; Nishimura, Y.; Sawada, T.; Kawafune, H.; Tsumoto, H.; Tsuji, Y.; Zhao, J.; Kyotani, Y.; Tanaka, T.; et al. S-nitrosylation regulates mitochondrial quality control via activation of parkin. Sci. Rep. 2013, 3, 2202.
- Piantadosi, C.A. Regulation of mitochondrial processes by protein S-nitrosylation. Biochim. Biophys. Acta 2012, 1820, 712–721.
- Ghafourifar, P.; Cadenas, E. Mitochondrial nitric oxide synthase. Trends. Pharm. Sci. 2005, 26, 190–195.
- Zhang, J.; Jin, B.; Li, L.; Block, E.R.; Patel, J.M. Nitric oxide-induced persistent inhibition and nitrosylation of active site cysteine residues of mitochondrial cytochrome-c oxidase in lung endothelial cells. Am. J. Physiol. Cell Physiol. 2005, 288, C840–C849.
- Brown, G.C.; Borutaite, V. Inhibition of mitochondrial respiratory complex I by nitric oxide, peroxynitrite and S-nitrosothiols. Biochim. Biophys. Acta 2004, 1658, 44–49.
- Hagen, T.; Taylor, C.T.; Lam, F.; Moncada, S. Redistribution of intracellular oxygen in hypoxia by nitric oxide: Effect on HIF1alpha. Science 2003, 302, 1975–1978.
- Chang, A.H.; Sancheti, H.; Garcia, J.; Kaplowitz, N.; Cadenas, E.; Han, D. Respiratory substrates regulate S-nitrosylation of mitochondrial proteins through a thiol-dependent pathway. Chem. Res. Toxicol. 2014, 27, 794–804.
- Zorova, L.D.; Popkov, V.A.; Plotnikov, E.Y.; Silachev, D.N.; Pevzner, I.B.; Jankauskas, S.S.; Babenko, V.A.; Zorov, S.D.; Balakireva, A.V.; Juhaszova, M.; et al. Mitochondrial membrane potential. Anal. Biochem. 2018, 552, 50–59.
- Basu, S.; Keszler, A.; Azarova, N.A.; Nwanze, N.; Perlegas, A.; Shiva, S.; Broniowska, K.A.; Hogg, N.; Kim-Shapiro, D.B. A novel role for cytochrome c: Efficient catalysis of S-nitrosothiol formation. Free Radic. Biol. Med. 2010, 48, 255–263.
- Schonhoff, C.M.; Gaston, B.; Mannick, J.B. Nitrosylation of cytochrome c during apoptosis. J. Biol. Chem. 2003, 278, 18265–18270.
- Speijer, D.; Manjeri, G.R.; Szklarczyk, R. How to deal with oxygen radicals stemming from mitochondrial fatty acid oxidation. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2014, 369, 20130446.
- Quijano, C.; Trujillo, M.; Castro, L.; Trostchansky, A. Interplay between oxidant species and energy metabolism. Redox. Biol. 2016, 8, 28–42.
- Quinlan, C.L.; Perevoshchikova, I.V.; Hey-Mogensen, M.; Orr, A.L.; Brand, M.D. Sites of reactive oxygen species generation by mitochondria oxidizing different substrates. Redox. Biol. 2013, 1, 304–312.
- Seim, G.L.; John, S.V.; Arp, N.L.; Fang, Z.; Pagliarini, D.J.; Fan, J. Nitric oxide-driven modifications of lipoic arm inhibit alpha-ketoacid dehydrogenases. Nat. Chem. Biol. 2022. Online ahead of print.
- Lemasters, J.J.; Theruvath, T.P.; Zhong, Z.; Nieminen, A.L. Mitochondrial calcium and the permeability transition in cell death. Biochim. Biophys. Acta 2009, 1787, 1395–1401.
- Nguyen, T.T.; Stevens, M.V.; Kohr, M.; Steenbergen, C.; Sack, M.N.; Murphy, E. Cysteine 203 of cyclophilin D is critical for cyclophilin D activation of the mitochondrial permeability transition pore. J. Biol. Chem. 2011, 286, 40184–40192.
- Ohtani, H.; Katoh, H.; Tanaka, T.; Saotome, M.; Urushida, T.; Satoh, H.; Hayashi, H. Effects of nitric oxide on mitochondrial permeability transition pore and thiol-mediated responses in cardiac myocytes. Nitric. Oxide. 2012, 26, 95–101.
- Shoshan-Barmatz, V.; De Pinto, V.; Zweckstetter, M.; Raviv, Z.; Keinan, N.; Arbel, N. VDAC, a multi-functional mitochondrial protein regulating cell life and death. Mol. Asp. Med. 2010, 31, 227–285.
- Cheng, Q.; Sedlic, F.; Pravdic, D.; Bosnjak, Z.J.; Kwok, W.M. Biphasic effect of nitric oxide on the cardiac voltage-dependent anion channel. FEBS Lett. 2011, 585, 328–334.
- Mannick, J.B.; Schonhoff, C.; Papeta, N.; Ghafourifar, P.; Szibor, M.; Fang, K.; Gaston, B. S-Nitrosylation of mitochondrial caspases. J. Cell Biol. 2001, 154, 1111–1116.
- Crow, M.T.; Mani, K.; Nam, Y.J.; Kitsis, R.N. The mitochondrial death pathway and cardiac myocyte apoptosis. Circ. Res. 2004, 95, 957–970.
- Azad, N.; Vallyathan, V.; Wang, L.; Tantishaiyakul, V.; Stehlik, C.; Leonard, S.S.; Rojanasakul, Y. S-nitrosylation of Bcl-2 inhibits its ubiquitin-proteasomal degradation. A novel antiapoptotic mechanism that suppresses apoptosis. J. Biol. Chem. 2006, 281, 34124–34134.
- Gu, Z.; Nakamura, T.; Lipton, S.A. Redox reactions induced by nitrosative stress mediate protein misfolding and mitochondrial dysfunction in neurodegenerative diseases. Mol. Neurobiol. 2010, 41, 55–72.
- Nakamura, T.; Cieplak, P.; Cho, D.H.; Godzik, A.; Lipton, S.A. S-nitrosylation of Drp1 links excessive mitochondrial fission to neuronal injury in neurodegeneration. Mitochondrion 2010, 10, 573–578.

