 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Gonzalo I Gómez | -- | 2603 | 2022-12-22 14:38:12 | | | |
| 2 | Gonzalo I Gómez | Meta information modification | 2603 | 2022-12-22 14:39:43 | | | | |
| 3 | Vivi Li | Meta information modification | 2603 | 2022-12-23 03:59:53 | | |
Video Upload Options
Hypertension is one of the most common risk factors for developing chronic cardiovascular diseases, including hypertensive nephropathy. Within the glomerulus, hypertension causes damage and activation of mesangial cells (MCs), eliciting the production of large amounts of vasoactive and proinflammatory agents. Accordingly, the activation of AT1 receptors by the vasoactive molecule angiotensin II (AngII) contributes to the pathogenesis of renal damage, which is mediated mostly by the dysfunction of intracellular Ca2+ ([Ca2+]i) signaling. Similarly, inflammation entails complex processes, where [Ca2+]i also play crucial roles. Deregulation of this second messenger increases cell damage and promotes fibrosis, reduces renal blood flow, and impairs the glomerular filtration barrier. In vertebrates, [Ca2+]i signaling depends, in part, on the activity of two families of large-pore channels: hemichannels and pannexons. Interestingly, the opening of these channels depends on [Ca2+]i signaling.
1. Introduction
1.1. Chronic Kidney Disease, General Aspects
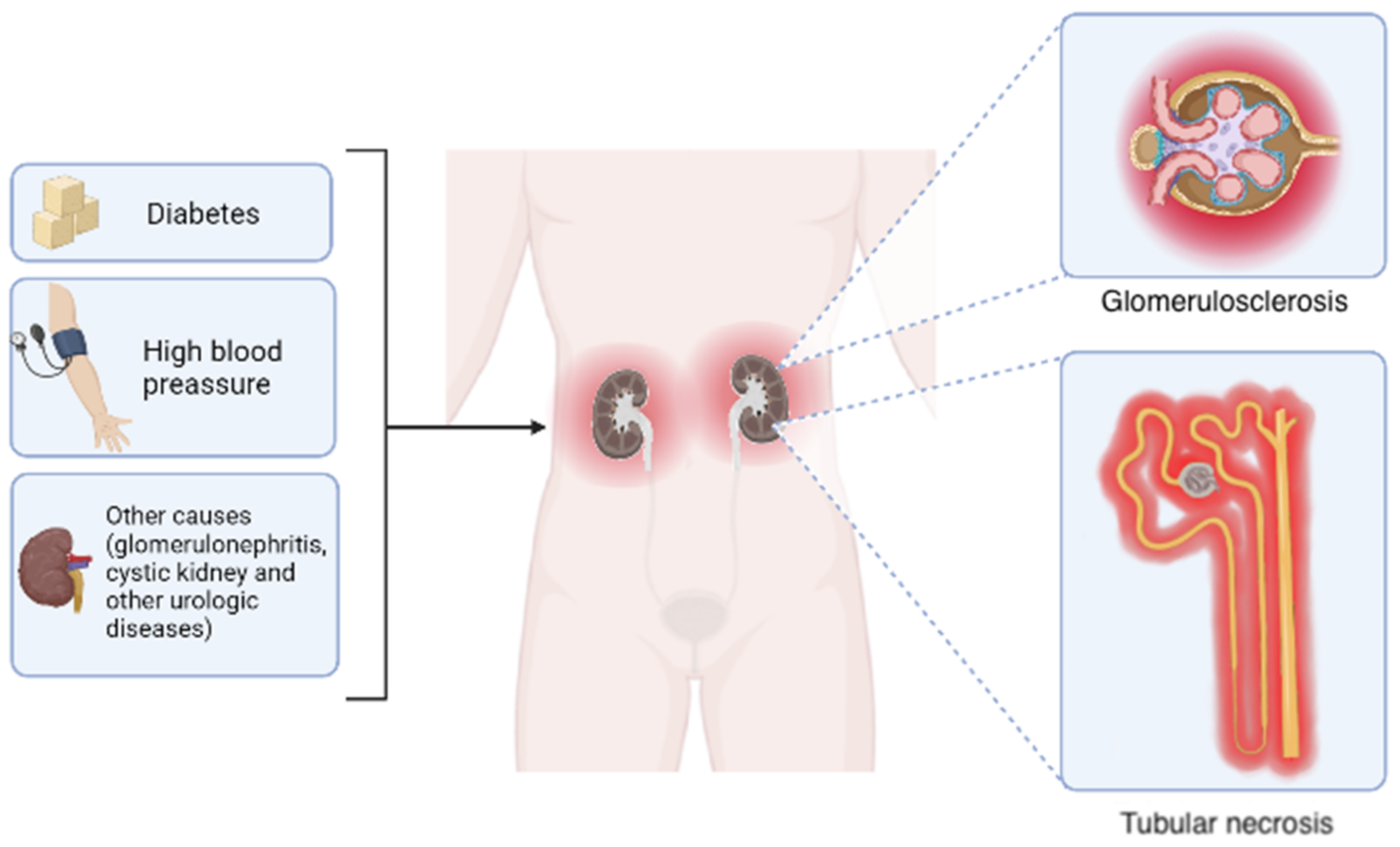
1.2. Role of Renin–Angiotensin System in CKD Development
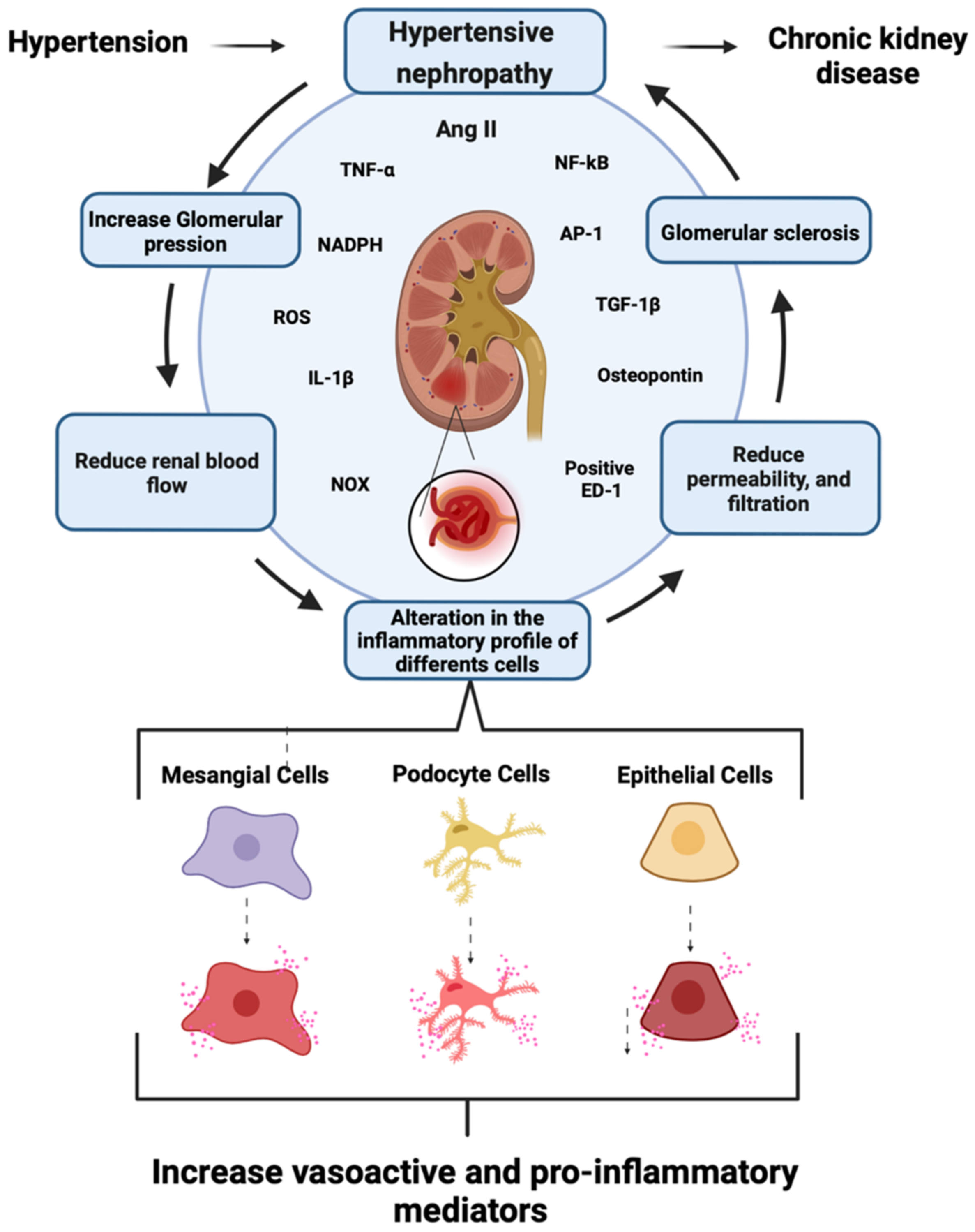
1.3. Role of Mesangial Cells in CKD
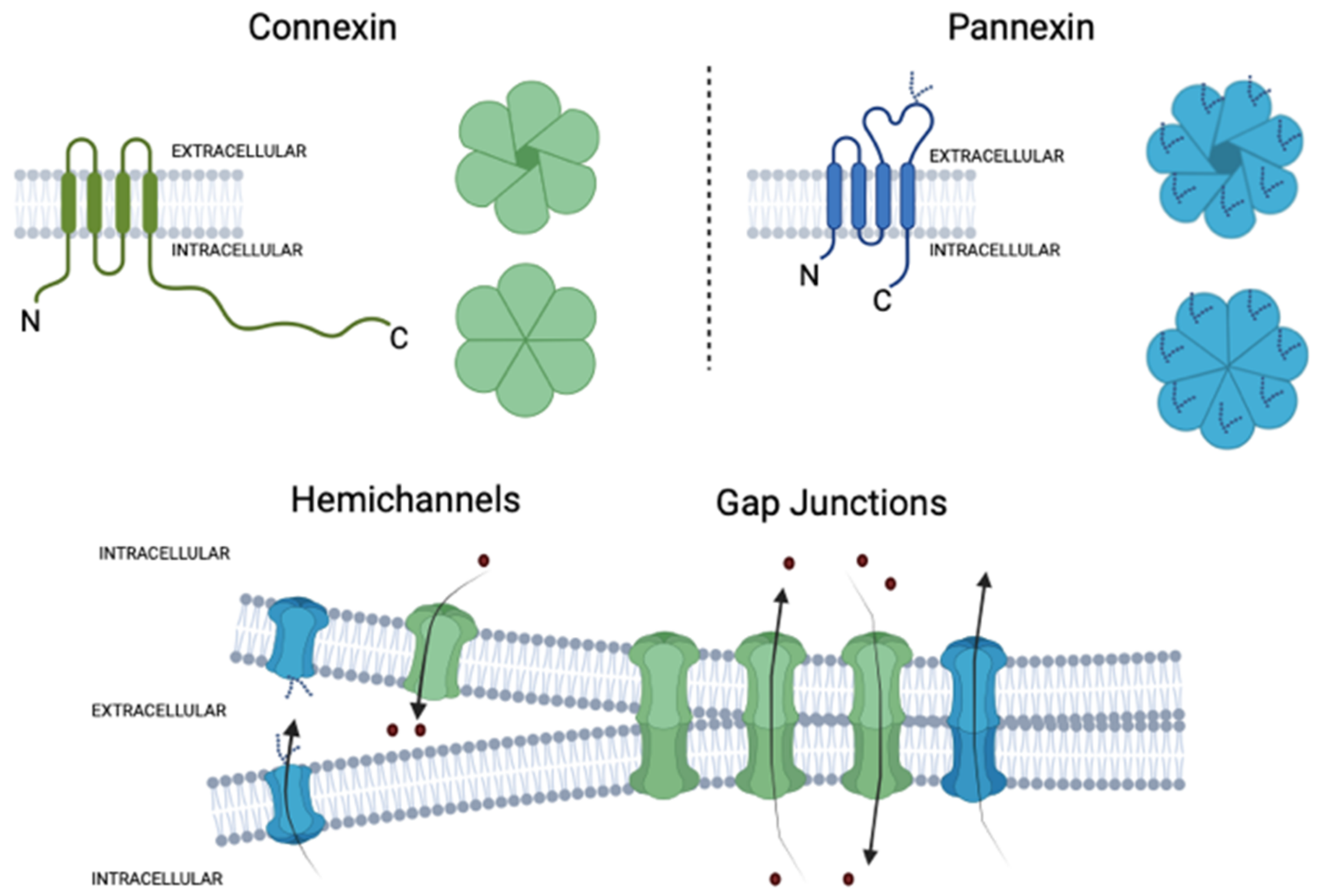
2. Connexins and Pannexins
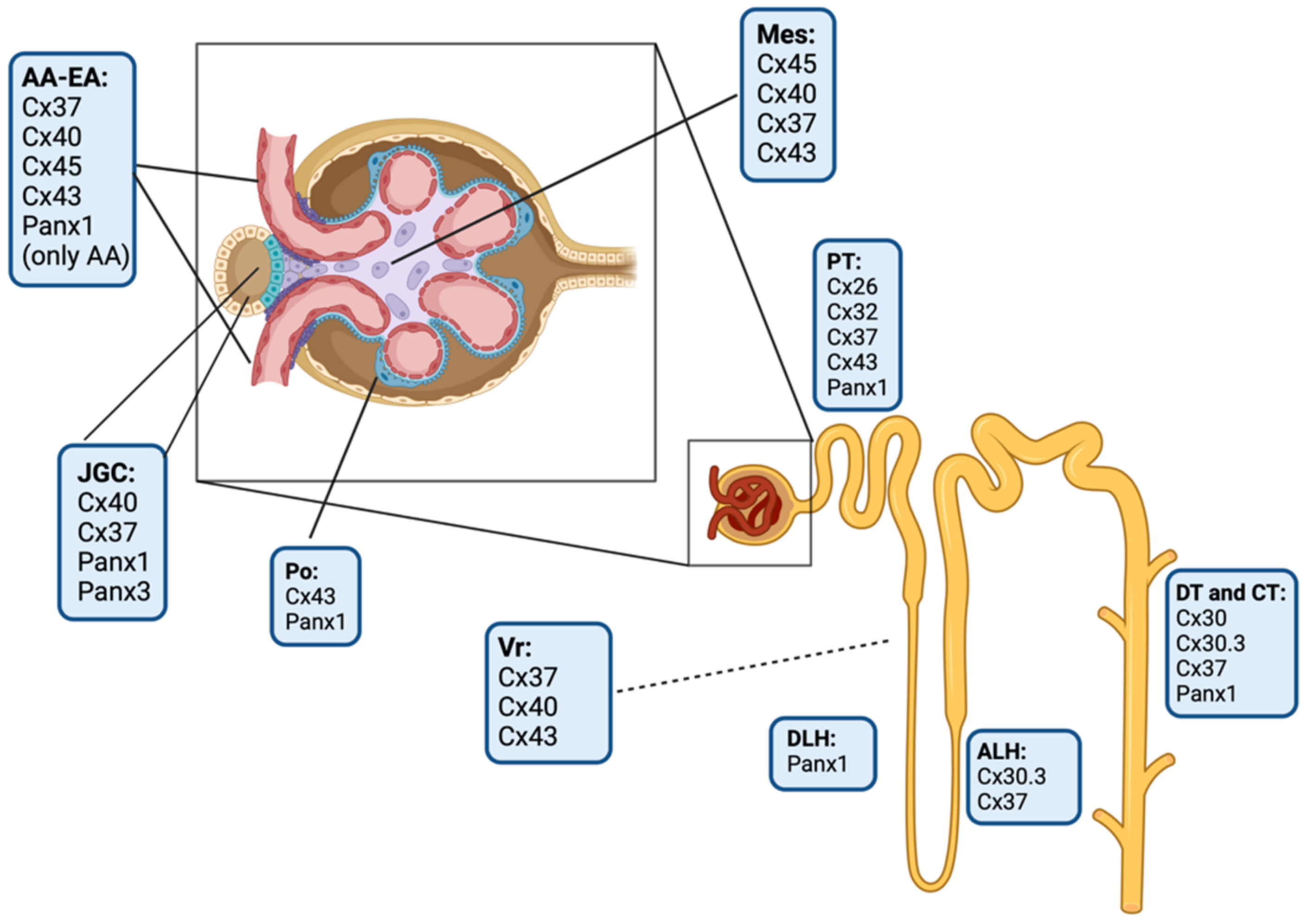
3. Connexins and Pannexin-Based Channels in the Kidney
References
- Atherton, J.C. Renal physiology. Br. J. Anaesth. 1972, 44, 236–245.
- Mohandas, R.; Douma, L.G.; Scindia, Y.; Gumz, M.L. Circadian Rhythms and Renal Pathophysiology. J. Clin. Investig. 2022, 132, e148277.
- Brown, D.; Bouley, R.; Păunescu, T.; Breton, S.; Lu, H. New Insights into the Dynamic Regulation of Water and Acid-Base Balance by Renal Epithelial Cells.o Title. Am. J. Physiol. Cell Physiol. 2012, 302, C1421–C1433.
- Lv, J.-C.; Zhang, L.-X. Prevalence and Disease Burden of Chronic Kidney Disease. In Renal Fibrosis: Mechanisms and Therapies; Springer: Berlin, Germany, 2019; pp. 3–15.
- de Zeeuw, D.; Hillege, H.L.; de Jong, P.E. The Kidney, a Cardiovascular Risk Marker, and a New Target for Therapy. Kidney Int. Suppl. 2005, 68, S25–S29.
- Gómez, G.I.; Velarde, V. Boldine Improves Kidney Damage in the Goldblatt 2K1C Model Avoiding the Increase in TGF-β. Int. J. Mol. Sci. 2018, 19, 1864.
- Lv, J.C.; Zhang, L.X. Prevalence and Disease Burden of Chronic Kidney Disease; Springer: Singapore, 2019; Volume 1165, ISBN 9789811388712.
- Bikbov, B.; Purcell, C.A.; Levey, A.S.; Smith, M.; Abdoli, A.; Abebe, M.; Adebayo, O.M.; Afarideh, M.; Agarwal, S.K.; Agudelo-Botero, M.; et al. Global, Regional, and National Burden of Chronic Kidney Disease, 1990–2017: A Systematic Analysis for the Global Burden of Disease Study 2017. Lancet 2020, 395, 709–733.
- Coresh, J.; Byrd-Holt, D.; Astor, B.C.; Briggs, J.P.; Eggers, P.W.; Lacher, D.A.; Hostetter, T.H. Chronic Kidney Disease Awareness, Prevalence, and Trends among U.S. Adults, 1999 to 2000. J. Am. Soc. Nephrol. 2004, 16, 180–188.
- Nangaku, M. Chronic Hypoxia and Tubulointerstitial Injury: A Final Common Pathway to End-Stage Renal Failure. J. Am. Soc. Nephrol. 2005, 17, 17–25.
- Gómez, G.I.; Velarde, V.; Sáez, J.C. Connexin-Based Channels and RhoA/ROCK Pathway in Angiotensin II-Induced Kidney Damage. In Selected Chapters from the Renin-Angiotensin System; IntechOpen: London, UK, 2020.
- Fleck, C.; Appenroth, D.; Jonas, P.; Koch, M.; Kundt, G.; Nizze, H.; Stein, G. Suitability of 5/6 Nephrectomy (5/6NX) for the Induction of Interstitial Renal Fibrosis in Rats--Influence of Sex, Strain, and Surgical Procedure. Exp. Toxicol. Pathol. 2006, 57, 195–205.
- Singri, N.; Ahya, S.N.; Levin, M.L. Acute Renal Failure. JAMA 2003, 289, 747–751.
- Levey, A.S.; Coresh, J.; Balk, E.; Kausz, A.T.; Levin, A.; Steffes, M.W.; Hogg, R.J.; Perrone, R.D.; Lau, J.; Eknoyan, G.; et al. National Kidney Foundation Practice Guidelines for Chronic Kidney Disease: Evaluation, Classification, and Stratification. Ann. Intern. Med. 2003, 139, 137–147.
- Gómez, G.; Fernández, P.; Velarde, V.; Sáez, J. Angiotensin II-Induced Mesangial Cell Damage Is Preceded by Cell Membrane Permeabilization Due to Upregulation of Non-Selective Channels. Int. J. Mol. Sci. 2018, 19, 957.
- López-Novoa, J.M.; Martínez-Salgado, C.; Rodríguez-Peña, A.B.; López-Hernández, F.J. Common Pathophysiological Mechanisms of Chronic Kidney Disease: Therapeutic Perspectives. Pharmacol. Ther. 2010, 128, 61–81.
- Petreski, T.; Piko, N.; Ekart, R.; Hojs, R.; Bevc, S. Review on Inflammation Markers in Chronic Kidney Disease. Biomedicines 2021, 9, 182.
- Rosivall, L. Intrarenal Renin–Angiotensin System. Mol. Cell Endocrinol. 2009, 302, 185–192.
- Seikaly, M.G.; Arant, B.S.; Seney, F.D. Endogenous Angiotensin Concentrations in Specific Intrarenal Fluid Compartments of the Rat. J. Clin. Investig. 1990, 86, 1352–1357.
- Rightsel, W.A.; Okamura, T.; Inagami, T.; Pitcock, J.A.; Takii, Y.; Brooks, B.; Brown, P.; Muirhead, E.E. Juxtaglomerular Cells Grown as Monolayer Cell Culture Contain Renin, Angiotensin I-Converting Enzyme, and Angiotensin I and II/III. Circ. Res. 1982, 50, 822–829.
- Costantino, V.V.; Gil Lorenzo, A.F.; Bocanegra, V.; Vallés, P.G. Molecular Mechanisms of Hypertensive Nephropathy: Renoprotective Effect of Losartan through Hsp70. Cells 2021, 10, 3146.
- Border, W.A.; Noble, N.A. Interactions of Transforming Growth Factor-Beta and Angiotensin II in Renal Fibrosis. Hypertension 1998, 31, 181–188.
- Sanchez-Lopez, E.; Rodriguez-Vita, J.; Cartier, C.; Rupérez, M.; Esteban, V.; Carvajal, G.; Díez, R.R.; Plaza, J.J.; Egido, J.; Ruiz-Ortega, M. Inhibitory effect of interleukin-1β on angiotensin II-induced connective tissue growth factor and type IV collagen production in cultured mesangial cells. Am. J. Physiol. Physiol. 2008, 294, F149–F160.
- Singh, P.; Bahrami, L.; Castillo, A.; Majid, D.S.A. TNF-α Type 2 Receptor Mediates Renal Inflammatory Response to Chronic Angiotensin II Administration with High Salt Intake in Mice. Am. J. Physiol. Ren. Physiol. 2013, 304, F991–F999.
- Mezzano, S.A.; Ruiz-Ortega, M.; Egido, J. Angiotensin II and Renal Fibrosis. Hypertension 2001, 38, 635–638.
- Mezzano, S.A.; Aros, C.A.; Droguett, A.; Burgos, M.E.; Ardiles, L.G.; Flores, C.A.; Carpio, D.; Vío, C.P.; Ruiz-Ortega, M. Jes Renal angiotensin II up-regulation and myofibroblast activation in human membranous nephropathy. Kidney Int. 2003, 64, S39–S45.
- Ozawa, Y.; Kobori, H.; Suzaki, Y.; Navar, L.G. Sustained renal interstitial macrophage infiltration following chronic angiotensin II infusions. Am. J. Physiol. Physiol. 2007, 292, F330–F339.
- Rivera, P.; Ocaranza, M.P.; Lavandero, S.; Jalil, J.E. Rho Kinase Activation and Gene Expression Related to Vascular Remodeling in Normotensive Rats with High Angiotensin I Converting Enzyme Levels. Hypertension 2007, 50, 792–798.
- Zhao, W.; Chen, S.S.; Chen, Y.; Ahokas, R.A.; Sun, Y. Kidney Fibrosis in Hypertensive Rats: Role of Oxidative Stress. Am. J. Nephrol. 2008, 28, 548–554.
- Clermont, G.; Lecour, S.; Lahet, J.-J.; Siohan, P.; Vergely, C.; Chevet, D.; Rifle, G.; Rochette, L. Alteration in plasma antioxidant capacities in chronic renal failure and hemodialysis patients: A possible explanation for the increased cardiovascular risk in these patients. Cardiovasc. Res. 2000, 47, 618–623.
- Johnson, R.J.; Alpers, C.E.; Yoshimura, A.; Lombardi, D.; Pritzl, P.; Floege, J.; Schwartz, S.M. Renal Injury from Angiotensin II-Mediated Hypertension. Hypertension 1992, 19, 464–474.
- Lohmeier, T.E. Angiotensin II Infusion Model of Hypertension. Hypertension 2012, 59, 539–541.
- Hernández-Salinas, R.; Vielma, A.Z.; Arismendi, M.N.; Boric, M.P.; Sáez, J.C.; Velarde, V. Boldine Prevents Renal Alterations in Diabetic Rats. J. Diabetes Res. 2013, 2013, 593672.
- Herrera, G.A. Plasticity of Mesangial Cells: A Basis for Understanding Pathological Alterations. Ultrastruct. Pathol. 2006, 30, 471–479.
- Sequeira-Lopez, M.L.S.; Gomez, R.A. Renin Cells, the Kidney, and Hypertension. Circ. Res. 2021, 128, 887–907.
- Moon, J.-Y.; Tanimoto, M.; Gohda, T.; Hagiwara, S.; Yamazaki, T.; Ohara, I.; Murakoshi, M.; Aoki, T.; Ishikawa, Y.; Lee, S.-H.; et al. Attenuating effect of angiotensin-(1–7) on angiotensin II-mediated NAD(P)H oxidase activation in type 2 diabetic nephropathy of KK-Ay/Ta mice. Am. J. Physiol. Physiol. 2011, 300, F1271–F1282.
- Yue, L.; Wang, W.; Wang, Y.; Du, T.; Shen, W.; Tang, H.; Wang, Y.; Yin, H. Bletilla Striata Polysaccharide Inhibits Angiotensin II-Induced ROS and Inflammation via NOX4 and TLR2 Pathways. Int. J. Biol. Macromol. 2016, 89, 376–388.
- Ding, G.-X.; Zhang, A.-H.; Huang, S.-M.; Pan, X.-Q.; Chen, R.-H. SP600125, an inhibitor of c-Jun NH2-terminal kinase, blocks expression of angiotensin II-induced monocyte chemoattractant protein-1 in human mesangial cells. World J. Pediatr. 2010, 6, 169–176.
- Bolick, D.T.; Hatley, M.E.; Srinivasan, S.; Hedrick, C.C.; Nadler, J.L. Lisofylline, a Novel Antiinflammatory Compound, Protects Mesangial Cells from Hyperglycemia- and Angiotensin II-Mediated Extracellular Matrix Deposition. Endocrinology 2003, 144, 5227–5231.
- Söhl, G.; Maxeiner, S.; Willecke, K. Expression and functions of neuronal gap junctions. Nat. Rev. Neurosci. 2005, 6, 191–200.
- Goodenough, D.A.; Paul, D.L. Gap Junctions. Cold Spring Harb. Perspect. Biol. 2009, 1, a002576.
- Nielsen, M.S.; Nygaard Axelsen, L.; Sorgen, P.L.; Verma, V.; Delmar, M.; Holstein-Rathlou, N. Gap Junctions. In Comprehensive Physiology; Wiley: New York, NY, USA, 2012; pp. 1981–2035.
- Scemes, E.; Spray, D.C.; Meda, P. Connexins, Pannexins, Innexins: Novel Roles of “Hemi-Channels”. Pflug. Arch. 2009, 457, 1207–1226.
- Leybaert, L.; Lampe, P.D.; Dhein, S.; Kwak, B.R.; Ferdinandy, P.; Beyer, E.C.; Laird, D.W.; Naus, C.C.; Green, C.R.; Schulz, R. Connexins in Cardiovascular and Neurovascular Health and Disease: Pharmacological Implications. Pharm. Rev. 2017, 69, 396–478.
- Nielsen, B.S.; Alstrom, J.S.; Nicholson, B.J.; Nielsen, M.S.; MacAulay, N. Permeant-Specific Gating of Connexin 30 Hemichannels. J. Biol. Chem. 2017, 292, 19999–20009.
- Hansen, D.B.; Ye, Z.-C.; Calloe, K.; Braunstein, T.H.; Hofgaard, J.P.; Ransom, B.R.; Nielsen, M.S.; MacAulay, N. Activation, Permeability, and Inhibition of Astrocytic and Neuronal Large Pore (Hemi)Channels. J. Biol. Chem. 2014, 289, 26058–26073.
- Hansen, D.B.; Braunstein, T.H.; Nielsen, M.S.; MacAulay, N. Distinct Permeation Profiles of the Connexin 30 and 43 Hemichannels. FEBS Lett. 2014, 588, 1446–1457.
- Nielsen, B.S.; Zonta, F.; Farkas, T.; Litman, T.; Nielsen, M.S.; MacAulay, N. Structural Determinants Underlying Permeant Discrimination of the Cx43 Hemichannel. J. Biol. Chem. 2019, 294, 16789–16803.
- Beblo, D.A.; Veenstra, R.D. Monovalent Cation Permeation through the Connexin40 Gap Junction Channel. J. Gen. Physiol. 1997, 109, 509–522.
- Suchyna, T.M.; Nitsche, J.M.; Chilton, M.; Harris, A.L.; Veenstra, R.D.; Nicholson, B.J. Different Ionic Selectivities for Connexins 26 and 32 Produce Rectifying Gap Junction Channels. Biophys. J. 1999, 77, 2968–2987.
- Wang, H.-Z.; Veenstra, R.D. Monovalent Ion Selectivity Sequences of the Rat Connexin43 Gap Junction Channel. J. Gen. Physiol. 1997, 109, 491–507.
- Goldberg, G.S.; Moreno, A.P.; Lampe, P.D. Gap Junctions between Cells Expressing Connexin 43 or 32 Show Inverse Permselectivity to Adenosine and ATP. J. Biol. Chem. 2002, 277, 36725–36730.
- Belliveau, D.J.; Bani-Yaghoub, M.; McGirr, B.; Naus, C.C.G.; Rushlow, W.J. Enhanced Neurite Outgrowth in PC12 Cells Mediated by Connexin Hemichannels and ATP. J. Biol. Chem. 2006, 281, 20920–20931.
- Sarieddine, M.Z.R.; Scheckenbach, K.E.L.; Foglia, B.; Maass, K.; Garcia, I.; Kwak, B.R.; Chanson, M. Connexin43 Modulates Neutrophil Recruitment to the Lung. J. Cell Mol. Med. 2009, 13, 4560–4570.
- Hanner, F.; Schnichels, M.; Zheng-Fischhöfer, Q.; Yang, L.E.; Toma, I.; Willecke, K.; McDonough, A.A.; Peti-Peterdi, J. Connexin 30.3 Is Expressed in the Kidney But Not Regulated by Dietary Salt or High Blood Pressure. Cell Commun. Adhes. 2008, 15, 219–230.
- Peracchia, C. Chemical gating of gap junction channels: Roles of calcium, pH and calmodulin. Biochim. Biophys. Acta (BBA) Biomembr. 2004, 1662, 61–80.
- Liu, S.; Taffet, S.; Stoner, L.; Delmar, M.; Vallano, M.L.; Jalife, J. A Structural Basis for the Unequal Sensitivity of the Major Cardiac and Liver Gap Junctions to Intracellular Acidification: The Carboxyl Tail Length. Biophys. J. 1993, 64, 1422–1433.
- Saez, J.C.; Spray, D.C.; Nairn, A.C.; Hertzberg, E.; Greengard, P.; Bennett, M. v CAMP Increases Junctional Conductance and Stimulates Phosphorylation of the 27-KDa Principal Gap Junction Polypeptide. Proc. Natl. Acad. Sci. USA 1986, 83, 2473–2477.
- Retamal, M.A.; Cortes, C.J.; Reuss, L.; Bennett, M.V.L.; Saez, J.C. S-Nitrosylation and Permeation through Connexin 43 Hemichannels in Astrocytes: Induction by Oxidant Stress and Reversal by Reducing Agents. Proc. Natl. Acad. Sci. USA 2006, 103, 4475–4480.
- Chandrasekhar, A.; Bera, A.K. Hemichannels: Permeants and Their Effect on Development, Physiology and Death. Cell Biochem. Funct. 2012, 30, 89–100.
- Sáez, J.C.; Schalper, K.A.; Retamal, M.A.; Orellana, J.A.; Shoji, K.F.; Bennett, M.V.L. Cell Membrane Permeabilization via Connexin Hemichannels in Living and Dying Cells. Exp. Cell Res. 2010, 316, 2377–2389.
- Retamal, M.A.; Reyes, E.P.; García, I.E.; Pinto, B.; Martínez, A.D.; González, C. Diseases associated with leaky hemichannels. Front. Cell. Neurosci. 2015, 9, 267.
- Prieto-Villalobos, J.; Alvear, T.F.; Liberona, A.; Lucero, C.M.; Martínez-Araya, C.J.; Balmazabal, J.; Inostroza, C.A.; Ramírez, G.; Gómez, G.I.; Orellana, J.A. Astroglial Hemichannels and Pannexons: The Hidden Link between Maternal Inflammation and Neurological Disorders. Int. J. Mol. Sci. 2021, 22, 9503.
- O’Carroll, S.J.; Alkadhi, M.; Nicholson, L.F.B.; Green, C.R. Connexin43 Mimetic Peptides Reduce Swelling, Astrogliosis, and Neuronal Cell Death after Spinal Cord Injury. Cell Commun. Adhes. 2008, 15, 27–42.
- Paul, D.L.; Ebihara, L.; Takemoto, L.J.; Swenson, K.I.; Goodenough, D.A. Connexin46, a Novel Lens Gap Junction Protein, Induces Voltage-Gated Currents in Nonjunctional Plasma Membrane of Xenopus Oocytes. J. Cell Biol. 1991, 115, 1077–1089.
- Fiori, M.C.; Figueroa, V.; Zoghbi, M.E.; Saéz, J.C.; Reuss, L.; Altenberg, G.A. Permeation of Calcium through Purified Connexin 26 Hemichannels. J. Biol. Chem. 2012, 287, 40826–40834.
- Schalper, K.A.; Sánchez, H.A.; Lee, S.C.; Altenberg, G.A.; Nathanson, M.H.; Sáez, J.C. Connexin 43 Hemichannels Mediate the Ca 2+ Influx Induced by Extracellular Alkalinization. Am. J. Physiol. Cell Physiol. 2010, 299, C1504–C1515.
- Kim, J.-C.; Pérez-Hernández, M.; Alvarado, F.J.; Maurya, S.R.; Montnach, J.; Yin, Y.; Zhang, M.; Lin, X.; Vasquez, C.; Heguy, A.; et al. Disruption of Ca 2+ i Homeostasis and Connexin 43 Hemichannel Function in the Right Ventricle Precedes Overt Arrhythmogenic Cardiomyopathy in Plakophilin-2–Deficient Mice. Circulation 2019, 140, 1015–1030.
- Yi, C.; Mei, X.; Ezan, P.; Mato, S.; Matias, I.; Giaume, C.; Koulakoff, A. Astroglial connexin43 contributes to neuronal suffering in a mouse model of Alzheimer’s disease. Cell Death Differ. 2016, 23, 1691–1701.
- Orellana, J.A.; Shoji, K.F.; Abudara, V.; Ezan, P.; Amigou, E.; Saez, P.J.; Jiang, J.X.; Naus, C.C.; Saez, J.C.; Giaume, C. Amyloid -Induced Death in Neurons Involves Glial and Neuronal Hemichannels. J. Neurosci. 2011, 31, 4962–4977.
- Barbe, M.T.; Monyer, H.; Bruzzone, R. Cell-Cell Communication Beyond Connexins: The Pannexin Channels. Physiology 2006, 21, 103–114.
- Penuela, S.; Harland, L.; Simek, J.; Laird, D.W. Pannexin Channels and Their Links to Human Disease. Biochem. J. 2014, 461, 371–381.
- Lohman, A.W.; Isakson, B.E. Differentiating Connexin Hemichannels and Pannexin Channels in Cellular ATP Release. FEBS Lett. 2014, 588, 1379–1388.
- Gajardo-Gómez, R.; Labra, V.C.; Orellana, J.A. Connexins and Pannexins: New Insights into Microglial Functions and Dysfunctions. Front. Mol. Neurosci. 2016, 9, 86.
- O’Donnell, B.L.; Penuela, S. Pannexin 3 Channels in Health and Disease. Purinergic Signal. 2021, 17, 577–589.
- Taylor, K.A.; Wright, J.R.; Mahaut-Smith, M.P. Regulation of Pannexin-1 Channel Activity. Biochem. Soc. Trans. 2015, 43, 502–507.
- Michalski, K.; Syrjanen, J.L.; Henze, E.; Kumpf, J.; Furukawa, H.; Kawate, T. The Cryo-EM Structure of Pannexin 1 Reveals Unique Motifs for Ion Selection and Inhibition. Elife 2020, 9, e54670.
- Palacios-Prado, N.; Soto, P.A.; López, X.; Choi, E.J.; Marquez-Miranda, V.; Rojas, M.; Duarte, Y.; Lee, J.; González-Nilo, F.D.; Sáez, J.C. Endogenous Pannexin1 Channels Form Functional Intercellular Cell–Cell Channels with Characteristic Voltage-Dependent Properties. Proc. Natl. Acad. Sci. USA 2022, 119, e2202104119.
- Billaud, M.; Sandilos, J.K.; Isakson, B.E. Pannexin 1 in the Regulation of Vascular Tone. Trends Cardiovasc. Med. 2012, 22, 68–72.
- Krick, S.; Wang, J.; St-Pierre, M.; Gonzalez, C.; Dahl, G.; Salathe, M. Dual Oxidase 2 (Duox2) Regulates Pannexin 1-Mediated ATP Release in Primary Human Airway Epithelial Cells via Changes in Intracellular PH and Not H2O2 Production. J. Biol. Chem. 2016, 291, 6423–6432.
- Chekeni, F.B.; Elliott, M.R.; Sandilos, J.K.; Walk, S.F.; Kinchen, J.M.; Lazarowski, E.R.; Armstrong, A.J.; Penuela, S.; Laird, D.W.; Salvesen, G.S.; et al. Pannexin 1 Channels Mediate “find-Me” Signal Release and Membrane Permeability during Apoptosis. Nature 2010, 467, 863–867.
- Adamson, S.E.; Leitinger, N. The Role of Pannexin1 in the Induction and Resolution of Inflammation. FEBS Lett. 2014, 588, 1416–1422.
- Silverman, W.R.; de Rivero Vaccari, J.P.; Locovei, S.; Qiu, F.; Carlsson, S.K.; Scemes, E.; Keane, R.W.; Dahl, G. The Pannexin 1 Channel Activates the Inflammasome in Neurons and Astrocytes. J. Biol. Chem. 2009, 284, 18143–18151.
- Su, L.; Jiang, X.; Yang, C.; Zhang, J.; Chen, B.; Li, Y.; Yao, S.; Xie, Q.; Gomez, H.; Murugan, R.; et al. Pannexin 1 Mediates Ferroptosis That Contributes to Renal Ischemia/Reperfusion Injury. J. Biol. Chem. 2019, 294, 19395–19404.
- Malik, S.; Eugenin, E.A. Role of Connexin and Pannexin Containing Channels in HIV Infection and NeuroAIDS. Neurosci. Lett. 2019, 695, 86–90.
- Penuela, S.; Gehi, R.; Laird, D.W. The Biochemistry and Function of Pannexin Channels. Biochim. Biophys. Acta (BBA) Biomembr. 2013, 1828, 15–22.
- Whyte-Fagundes, P.; Zoidl, G. Mechanisms of Pannexin1 Channel Gating and Regulation. Biochim. Biophys. Acta (BBA) Biomembr. 2018, 1860, 65–71.
- Volonté, C.; Apolloni, S.; Skaper, S.D.; Burnstock, G. P2X7 Receptors: Channels, Pores and More. CNS Neurol. Disord. Drug Targets 2012, 11, 705–721.
- Bao, L.; Locovei, S.; Dahl, G. Pannexin Membrane Channels Are Mechanosensitive Conduits for ATP. FEBS Lett. 2004, 572, 65–68.
- Locovei, S.; Wang, J.; Dahl, G. Activation of Pannexin 1 Channels by ATP through P2Y Receptors and by Cytoplasmic Calcium. FEBS Lett. 2006, 580, 239–244.
- Pinheiro, A.R.; Paramos-de-Carvalho, D.; Certal, M.; Costa, C.; Magalhães-Cardoso, M.T.; Ferreirinha, F.; Costa, M.A.; Correia-de-Sá, P. Bradykinin-Induced Ca2+ Signaling in Human Subcutaneous Fibroblasts Involves ATP Release via Hemichannels Leading to P2Y12 Receptors Activation. Cell Commun. Signal. 2013, 11, 70.
- Billaud, M.; Lohman, A.W.; Straub, A.C.; Looft-Wilson, R.; Johnstone, S.R.; Araj, C.A.; Best, A.K.; Chekeni, F.B.; Ravichandran, K.S.; Penuela, S.; et al. Pannexin1 Regulates A1-Adrenergic Receptor–Mediated Vasoconstriction. Circ. Res. 2011, 109, 80–85.
- Kurtz, A. Renal Connexins and Blood Pressure. Biochim. Biophys. Acta 2012, 1818, 1903–1908.
- Hanner, F.; Sorensen, C.M.; Holstein-Rathlou, N.-H.; Peti-Peterdi, J. Connexins and the kidney. Am. J. Physiol. Integr. Comp. Physiol. 2010, 298, R1143–R1155.
- Zhang, Q.; Cao, C.; Mangano, M.; Zhang, Z.; Silldorff, E.P.; Lee-Kwon, W.; Payne, K.; Pallone, T.L. Descending vasa recta endothelium is an electrical syncytium. Am. J. Physiol. Integr. Comp. Physiol. 2006, 291, R1688–R1699.
- Zehra, T.; Cupples, W.A.; Braam, B. Tubuloglomerular Feedback Synchronization in Nephrovascular Networks. J. Am. Soc. Nephrol. 2021, 32, 1293–1304.

