1. Introduction
1.1. Chronic Kidney Disease, General Aspects
The kidney is commonly described as an excretory organ and is essential to maintaining systemic homeostasis
[1][2][1,2]. Kidney-mediated blood filtering and modification of the ultrafiltration content by reabsorption and secretion, maintains pH, water, and electrolyte balance, contributing to regulating blood pressure and osmolality. The final product of the kidneys is urine, and its volume and composition can provide insights into body health status
[2]. As mentioned, the kidneys’ central role is to eliminate waste materials ingested or produced by metabolism and to control the fluid body volume and electrolyte composition
[3]. However, this organ can fail and when that happens, several deregulations in many organ functions can emerge. In this regard, chronic kidney disease (CKD) is a global health problem, the prevalence of which has increased
[4]. Around 10% of adults suffer kidney damage in developed countries
[5][6][5,6]. Currently, the estimated global prevalence of CKD is 697.5 million cases, and patients with end-stage renal disease (ESRD) who need renal replacement therapy range between 4.902 and 7.083 million
[7][8][7,8].
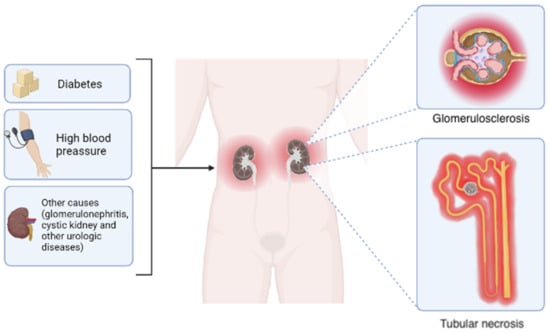
CKD embodies different etiologies, including diabetic nephropathy, hypertensive nephrosclerosis, and glomerulonephritis. However, regardless of the causes, some morphological changes are common, including tubular necrosis and glomerular sclerosis
[9][10][11][9,10,11] (
Figure 1). The pathogenesis of CKD begins with a disturbance of the glomerular and tubulointerstitial compartments due to the release of cytokines from the glomerulus. This local inflammatory impairs tubular epithelial cell functions, causing excessive protein filtration at the injured glomeruli
[11][12][11,12]. The above is then accompanied by tubulointerstitial ischemia at the glomerular lesion and the hyperfunction of the remaining tubules, promoting leukocyte, cytotoxicity, and fibrogenesis recruitment
[11][12][11,12] (
Figure 1). Altogether, these events could lead to acute renal failure, characterized by the abrupt and transitory decrease in renal function, inability to excrete waste products, and maintain the electrolyte balance, as well as generalized nephron dysfunction
[13]. In contrast, patients with CKD usually develop progressive kidney damage typified by glomerular sclerosis or tubulointerstitial fibrosis, which eventually leads to end-stage renal disease (ESRD), and the last stage of this condition causes irreversible damage
[14][15][14,15]. This phenomenon includes the progressive reduction in the glomerular filtration rate (GFR), due to increased nephron damage, which eventually triggers organ failure
[6][16][17][6,16,17].
Figure 1. Primary causes and morphological outcomes of chronic kidney disease. Although the leading causes of chronic kidney disease include diabetes and hypertension, other disorders such as glomerulonephritis, cystic kidney disease, and diverse urologic diseases also contribute in a minor proportion. Regardless of the etiology, the progressive reduction in glomerular filtration rate occurs accompanied by two common histological changes: glomerular sclerosis and tubular necrosis. Elaborated in
BioRender.com (accessed on 15 October 2022).
1.2. Role of Renin–Angiotensin System in CKD Development
Based on the renin localization, mainly in granular cells of the renal afferent arteriole, it is assumed that swelling or shrinking of the juxtaglomerular apparatus (JGA) is involved in the glomerular blood flow ascribed to the renin–angiotensin system (RAS). These cells make contact with the extraglomerular mesangium and macula densa, forming the JGA
[18]. The unique juxtaposition of the macula densa to the afferent arterioles suggests the existence of mechanisms linking tubular function with the control of arteriolar contraction
[18]. RAS is present not only in the juxtaglomerular apparatus and interstitium, but also in various segments of the nephron. Renal tubular RAS was recognized when the highest known angiotensin concentration in the body was measured in tubular fluid
[18][19][18,19]. Subsequently, it was shown that all components of RAS are present in vascular smooth muscle cells of afferent arterioles and glomerular podocytes, suggesting the involvement of the intracellular renin–angiotensin system in local regulatory systems
[18][20][18,20]. Hypertension is one of the most common risk factors for developing coronary artery disease, stroke, heart failure, peripheral arterial disease, vision loss, dementia, and CKD. The prevalence of hypertension among adults will escalate to more than 1.56 billion by 2025
[12][21][12,21]. Hypertensive nephropathy linked to hypertension is the second cause of terminal CKD, and, nowadays, the cost of its treatment exceeds the budget of health systems in many countries
[18][19][18,19]. The inflammatory injury and tissue remodeling evoked by CDK have been attributed to angiotensin II (Ang II), the principal peptide in RAS
[22].
AngII regulates blood pressure, intravascular volume, and electrolyte balance by acting on the kidneys and adrenal glands
[22]. Treatment with high concentrations of AngII induces the expression of chemokines, adhesion molecules, and pro-inflammatory cytokines (interleukin- 1β [IL-1β] and tumor necrosis factor-α [TNF-α])
[15][23][24][15,23,24]. The latter is accompanied by infiltration of macrophages (positive ED-1)
[25], and tubular overexpression of osteopontin, a macrophage chemoattractant and adhesion molecule
[25]. All these events are intimately associated with AngII-induced kidney damage linked to redox imbalance
[26][27][26,27]. AngII increases the expression of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (NOX), one of the essential enzymes in the generation of reactive oxygen species (ROS)
[28][29][28,29]. Indeed, the impairment of redox balance plays a decisive role in several chronic diseases, including the progression of CKD
[6][30][6,30] (
Figure 2). Given that intrarenal RAS is critical for the pathophysiology of hypertensive nephropathy
[6][31][6,31], the administration of AngII has been used for modeling kidney damage. In addition to providing a clinically relevant model for systemic hypertension, exposure to AngII leads to progressive and chronic nephron damage that recapitulates events occurring in ESRD
[27][31][32][27,31,32]. Due to the diverse effects of RAS, RAS inhibitors (including blockers of angiotensin converting enzyme, angiotensin receptors, renin/prorenin receptors, and renin release) reduce blood pressure and increase renal blood flow and GFR, decreasing the progression of renal tissue damage
[17].
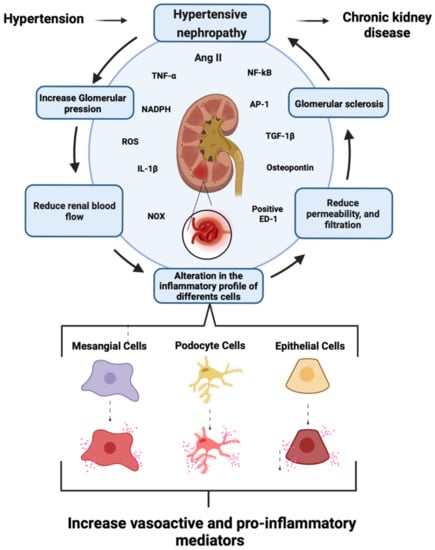
Figure 2. Schematics showing general aspects of the generation of chronic kidney disease mediated by hypertensive nephropathy. At the renal level, arterial hypertension increases intra-glomerular pressure, leading to reductions in renal blood flow and alterations in the inflammatory profile of mesangial cells, podocytes, and epithelial cells. The epithelial cells boost the production of vasoactive and pro-inflammatory mediators, triggering a reduction in glomerular permeability and filtration. Consequently, hypertensive nephropathy can contribute to CKD. Renal impairment would then be associated mainly with the persistent production of AngII, which could augment the expression of proinflammatory cytokines [IL-1β and TNF-α] and the NOX-mediated generation of ROS. These processes likely result in macrophage infiltration and tubular overexpression of osteopontin, favoring the production of TGF- β1 and activation of NF-κB and AP-1. Elaborated in
BioRender.com (accessed on 15 October 2022).
1.3. Role of Mesangial Cells in CKD
The renal corpuscle comprises the Bowman’s capsule and the glomerulus, both being fundamental structures for blood filtration. The Bowman’s capsule plays a vital role in the structural and functional stability of the glomerulus, allowing it to fulfill its filtering function successfully
[20][33][20,33]. Structurally speaking, the mesangium consists of a network of mesangial cells (MCs) that constitute between 30–40% of the glomerular cell populations. They control the capillary dilation, the regulation of the GFR, and the synthesis and degradation of extracellular matrix proteins
[33]. Moreover, they have contractile and phagocytic properties
[34]. Interestingly, cells from the renin lineage repopulate the niche of damaged glomerular MCs that have undergone mesangiolysis via the experimental model of mesangial proliferative glomerulonephritis. Still, the mechanism and molecules involved in the above phenomenon remain to be defined
[35]. On the other hand, MCs stimulated with AngII produce ROS
[14][36][37][14,36,37], synthesize and release IL-1β, TNF-α, and chemokines, such as the macrophage chemoattractant protein (MCP-1) and the transforming growth factor β1 (TGF-β1). These inflammatory mediators favor fibrosis, reducing renal blood flow, permeability, and eventually glomerular filtration
[14][20][27][33][37][38][39][14,20,27,33,37,38,39]. Transcription factors NF-κB and AP-1 command these alterations and increase the synthesis and release of extracellular matrix proteins, such as type IV collagen, laminin, and fibronectin (
Figure 2). The latter result in the formation of mesangial nodules and lesions at the interstitium, hindering the adequate function of the glomerulus
[27][33][37][38][39][27,33,37,38,39]. As MCs undergo progressive damage at the glomerulus, they may serve as a potential target for understanding the pathogenesis of chronic nephron lesions and subsequent CKD
[14][33][36][38][14,33,36,38].
2. Connexins and Pannexins
Gap junctions (GJs) are aggregates of ten to thousands of individual intercellular channels that permit the cytoplasmic exchange between adjacent cells. They allow the diffusion of ions, small metabolites (e.g., nicotinamide adenine dinucleotide: NAD+, glucose, lactate, and glutamate), and second messengers (e.g., cyclic adenosine monophosphate [cAMP] and inositol trisphosphate [IP3]) between adjacent cells
[40][41][42][40,41,42]. These connections support several biological processes in the animal kingdom, such as the propagation of electrical and chemical signals, which are crucial for cell coordination and survival. GJ channels are formed by integral membrane proteins called connexins, whose family has 21 isoforms in humans that are named according to their molecular weight
[42][43][42,43]. All connexins feature four transmembrane domains, two extracellular loops, each with three highly conserved cysteine residues, an intracellular loop, and the C- and N-terminal ends on the cytoplasmic domain
[42]. Six connexin monomers assemble around a central pore to form a hemichannel (HC) or connexons, whereas the apposition of two connexons from adjacent cells creates an intercellular GJ channel
[42]. Cells expressing more than one type of connexin could eventually form heteromeric connexons. Likewise, cells expressing different connexons could form heterotypic GJs
[42].
HCs act as free channels in the plasma membrane, favoring the exchange of small molecules and ions between the cytosol and the extracellular space
[43][44][43,44] (
Figure 3). Al-though initially they were catalogued as non-selective channels, nowadays, the evidence seems to indicate the opposite
[45][46][47][48][45,46,47,48]. For example, Cx32 HCs show a slight preference for anions, while Cx40 and Cx43 have a high selectivity for cations
[49][50][51][49,50,51]. In the same way, selectivity for ATP is 300-fold higher in Cx43 than in Cx32 HCs; for ADP, glutamate, and glutathione, the selectivity is 20-fold higher in Cx43 than in Cx32 HCs, while adenosine is 10-fold more permeable in Cx32 than Cx43 HCs
[42][52][42,52]. The physiological opening of these channels is critical for the proper function of multiple biological processes, such as neurite outgrowth
[53], neutrophil migration
[54], and regulation of acid–base homeostasis in the kidney
[55], among others. The HC opening is tightly regulated and relies on changes in voltage, intracellular/extracellular Ca
2+ concentration ([Ca
2+]
i)
[56], pH
[57], post-translational modifications (e.g., phosphorylation
[58] and S-nitrosylation
[59]), and protein interactions
[42]. Nevertheless, persistent activation of HCs has been associated with the genesis and progression of diverse pathological stages
[60][61][62][60,61,62]. At least three mechanisms have linked the exacerbated opening of HCs with cell dysfunction and damage
[63]. Firstly, the uncontrolled entry of Na
+ and Cl
− through HCs may result in osmotic and ionic imbalances coupled to further cell swelling and plasma membrane breakdown
[63][64][65][63,64,65]. Second, some HCs are permeable to Ca
2+ [66][67][66,67], which could permit its influx to the cytosol during pathological conditions. The direct or indirect increase in [Ca
2+]
i mediated by HCs could lead to Ca
2+ overload and consequent induction of different proteases, phospholipases, and other hydrolytic enzymes, as well as oxidative stress and caspase activation
[68][69][68,69]. Finally, exacerbated HC opening may trigger the release of high amounts of potentially toxic molecules to neighboring cells, such as glutamate, in the case of the brain
[63][70][63,70].
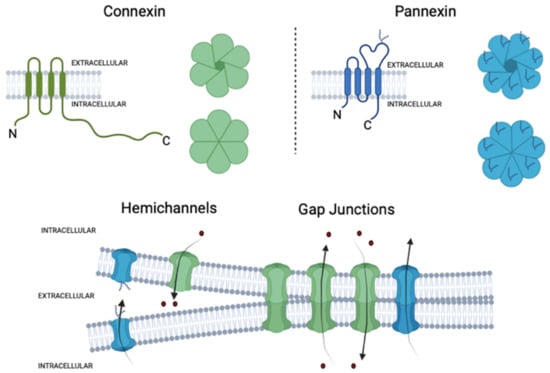
Figure 3. Structural organization of connexin and pannexin-based channels. Connexins and pannexins have four transmembrane domains (TM), two extracellular loops (EL), one cytoplasmatic loop (CL), and both the N- and C-terminus are localized on the intracellular side. The glycosylated extracellular loop of pannexins is also shown. Hemichannels or connexons are formed by the oligomerization of six subunits of connexins around a central pore, while pannexons are composed of seven pannexin subunits in the case of Panx1. Under normal conditions, the opening of hemichannels and pannexons is tightly regulated to fulfill several biological processes. Hemichannels and likely pannexons can also dock each other in junctional membrane interfaces to form cell-to-cell intercellular channels, termed gap junction channels. Elaborated in
BioRender.com (accessed on 15 October 2022).
Pannexins (Panxs) belong to a family of transmembrane proteins that bear structural homology with innexins, the GJ protein family of invertebrates
[71]. Although connexins and pannexins do not share significant sequence homology, both families exhibit similar transmembrane topology: four transmembrane domains with two extracellular loops and intracellular N- and C-terminals. Unlike connexins, which have several extracellular cysteine residues, pannexins have only two cysteines and a highly glycosylated aspartate in the second extracellular loop, which would hinder GJs formation (see below)
[72][73][72,73] (
Figure 3). Three pannexin isoforms have been identified to date: pannexin-1 (Panx1) (~48 kDa), is the most ubiquitous member of its family, broadly expressed in the brain, muscle, and other tissues
[74]; pannexin-2 (Panx2) (~45 kDa), predominantly expressed in the brain; and pannexin-3 (Panx3) (~73 kDa), found in skin, bone, cartilage, skeletal muscle, and blood vessels
[72][73][75][76][72,73,75,76]. Pannexin monomers oligomerize around a central pore in heptamers (Panx1, Panx3) or octamers (Panx2) called pannexons, allowing the exchange of ions and small molecules between the cytoplasm and the extracellular milieu
[71][72][77][71,72,77]. Panx1 channels have been historically described as non-junctional membrane channels; nevertheless, recent evidence suggests that Panx1 can form GJs channels due to a differential glycosylated state at its extracellular loop
[78]. Sequence analysis of Panx1 reveals that the Asp254 residue can be gradually glycosylated, which yields three different Panx1 bands in Western blot analysis: Gly-0 (non-glycosylated), Gly-1 (several mannose glycosylation), and Gly-2 (complex sugar glycosylation)
[78]. Mutagenesis studies show that these glycosylations facilitate Panx1 trafficking but are not essential for the arrival of Panx1 to the plasma membrane or the function of a GJ channel. However, the Gly-2 variant of Panx1 is predominant on the surface
[72][78][72,78].
Panx1 channels are associated with the regulation of vascular tone and flow
[79], mucociliary clearance in the airway epithelial
[80], phagocytosis of apoptotic cells
[81], activation of the inflammasome, and recruitment of immune cells
[82][83][82,83]. Similar to HCs, the persistent opening of Panx1 channels has been proposed to be harmful to cells, contributing to cell death in episodes of ischemia
[84] or facilitating viral infection
[85]. Panx1 channels have been extensively studied as crucial pathways for releasing ATP and pivotal players for purinergic receptor-mediated intracellular Ca
2+ wave propagation
[86][87][88][86,87,88]. Indeed, Panx1 channels release ATP at resting membrane potential and under physiological extracellular concentrations of divalent cations (Ca
2+ and Mg
2+)
[73]. Furthermore, multiple direct mechanisms activate these channels, including mechanical stimulation
[89], caspases-dependent cleavage of the C-tail of Panx1
[81], hypotonicity, ischemia
[73], or a secondary response to the activation of receptors ionotropic as P2X4, P2X7, and NMDAR, which are permeable to cations, including Na
+, K
+, and Ca
2+ [83][88][90][83,88,90], or metabotropic receptors such as P2Y1, P2Y2, and P2Y6
[90], protease-activated receptor-1 (PAR-1), bradykinin B
2 receptor (BDKRB2)
[91], histamine H
1 receptor (HRH1), and α
1D-adrenergic receptors (α
1D-AR)
[92].
3. Connexins and Pannexin-Based Channels in the Kidney
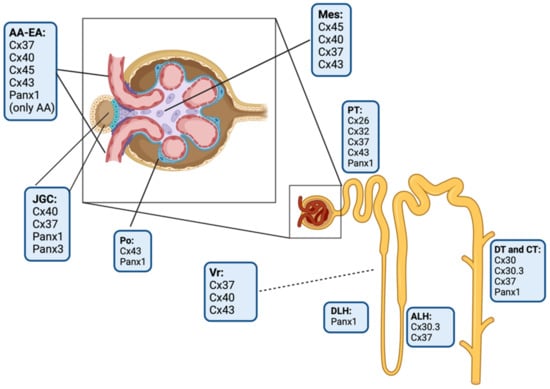
The kidney expresses nine connexin isoforms, including Cx26, Cx30, Cx30.3, Cx32, Cx37, Cx40, Cx43, Cx45, and Cx46
[93][94][95][96][93,94,95,96] (
Figure 4). The evidence indicates that these connexins could play a pivotal role in important aspects of renal function, such as the maintenance of acid–base homeostasis, hydroelectrolyte balance, regulation of blood pressure, and functional processes of the nephron: glomerular filtration, reabsorption and secretion of metabolites, water reabsorption, and renin secretion
[93][94][95][96][93,94,95,96].
Figure 4. Schematic drawing of the localization of connexin and pannexin isoforms in the kidney. AA, afferent arteriole; EA, efferent arteriole; JGC, juxtaglomerular cells; Po, podocytes; Mes, mesangial cells; PT, proximal tubules; DLH, descending limb of loop of Henle; ALH, ascending limb of loop of Henle; DT, distal tubules; CT, collecting tubules; and Vr, Vasa recta. Elaborated in
BioRender.com (accessed on 15 October 2022).