 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Jean-Louis Guéant | -- | 2760 | 2022-12-12 01:24:18 | | | |
| 2 | Conner Chen | + 33 word(s) | 2793 | 2022-12-12 11:08:21 | | | | |
| 3 | Conner Chen | -3 word(s) | 2790 | 2022-12-13 14:58:08 | | | | |
| 4 | Jean-Louis Guéant | Meta information modification | 2790 | 2023-01-12 17:19:47 | | |
Video Upload Options
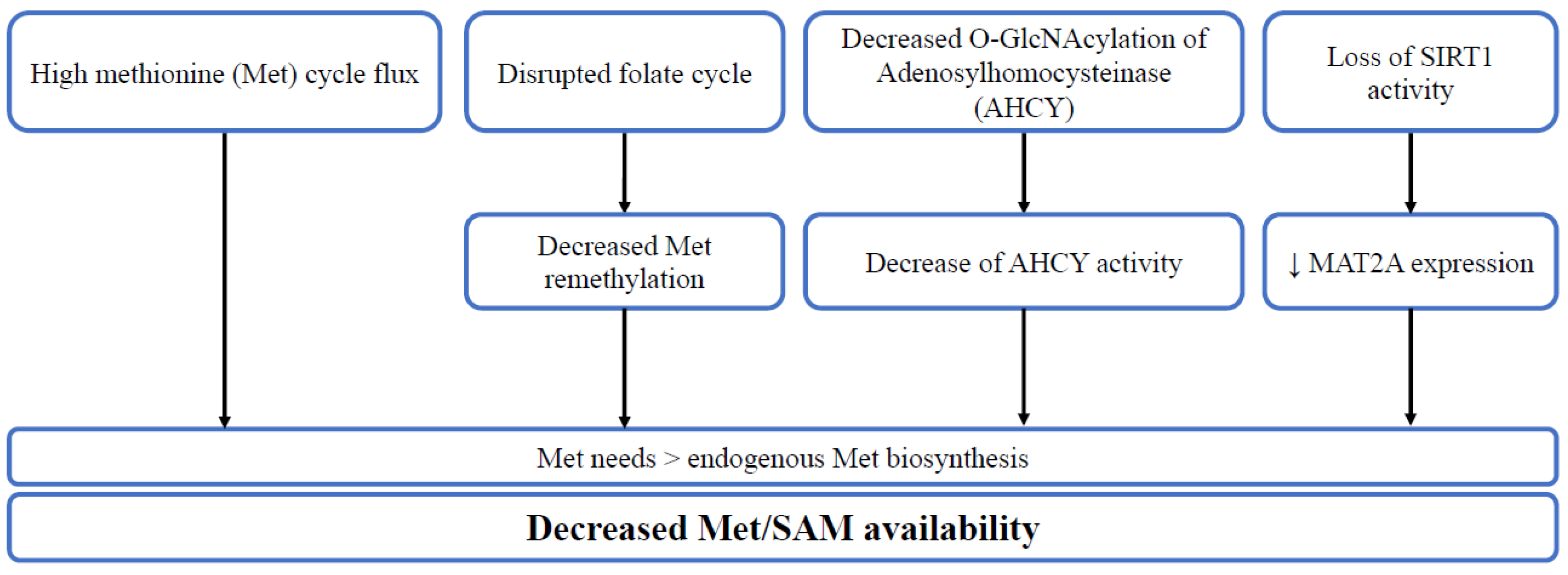
Stem cells are a population of undifferentiated cells with self-renewal and differentiation capacities. Normal and cancer stem cells share similar characteristics in relation to their stemness properties. One-carbon metabolism (OCM), a network of interconnected reactions, plays an important role in this dependence through its role in the endogenous synthesis of methionine and S-adenosylmethionine (SAM), the universal donor of methyl groups in eukaryotic cells. OCM genes are differentially expressed in stem cells, compared to their differentiated counterparts. Furthermore, cultivating stem cells in methionine-restricted conditions hinders their stemness capacities through decreased SAM levels with a subsequent decrease in histone methylation, notably H3K4me3, with a decrease in stem cell markers. Stem cells’ reliance on methionine is linked to several mechanisms, including high methionine flux or low endogenous methionine biosynthesis.
1. Methionine in Normal Stem Cells
2. Methionine in Cancer Stem Cells
3. The Influence of SIRT1 and PPAR-Alpha/PGC1-Alpha Pathway
References
- Wang, J.; Alexander, P.; Wu, L.; Hammer, R.; Cleaver, O.; McKnight, S.L. Dependence of mouse embryonic stem cells on threonine catabolism. Science 2009, 325, 435–439.
- Shiraki, N.; Shiraki, Y.; Tsuyama, T.; Obata, F.; Miura, M.; Nagae, G.; Aburatani, H.; Kume, K.; Endo, F.; Kume, S. Methionine metabolism regulates maintenance and differentiation of human pluripotent stem cells. Cell Metab. 2014, 19, 780–794.
- Kilberg, M.S.; Terada, N.; Shan, J. Influence of Amino Acid Metabolism on Embryonic Stem Cell Function and Differentiation. Adv. Nutr. 2016, 7, 780S–789S.
- Sim, E.Z.; Enomoto, T.; Shiraki, N.; Furuta, N.; Kashio, S.; Kambe, T.; Tsuyama, T.; Arakawa, A.; Ozawa, H.; Yokoyama, M.; et al. Methionine metabolism regulates pluripotent stem cell pluripotency and differentiation through zinc mobilization. Cell Rep. 2022, 40, 111120.
- Tang, S.; Fang, Y.; Huang, G.; Xu, X.; Padilla-Banks, E.; Fan, W.; Xu, Q.; Sanderson, S.M.; Foley, J.F.; Dowdy, S.; et al. Methionine metabolism is essential for SIRT1-regulated mouse embryonic stem cell maintenance and embryonic development. EMBO J. 2017, 36, 3175–3193.
- Zhu, Q.; Cheng, X.; Cheng, Y.; Chen, J.; Xu, H.; Gao, Y.; Duan, X.; Ji, J.; Li, X.; Yi, W. O-GlcNAcylation regulates the methionine cycle to promote pluripotency of stem cells. Proc. Natl. Acad. Sci. USA 2020, 117, 7755–7763.
- Hanover, J.A.; Krause, M.W.; Love, D.C. Bittersweet memories: Linking metabolism to epigenetics through O-GlcNAcylation. Nat. Rev. Mol. Cell Biol. 2012, 13, 312–321.
- Sheikh, M.A.; Emerald, B.S.; Ansari, S.A. Stem cell fate determination through protein O-GlcNAcylation. J. Biol. Chem. 2021, 296, 100035.
- Wells, L.; Vosseller, K.; Hart, G.W. Glycosylation of nucleocytoplasmic proteins: Signal transduction and O-GlcNAc. Science 2001, 291, 2376–2378.
- Sorin, M.; Watkins, D.; Gilfix, B.M.; Rosenblatt, D.S. Methionine dependence in tumor cells: The potential role of cobalamin and MMACHC. Mol. Genet. Metab. 2021, 132, 155–161.
- Kaiser, P. Methionine Dependence of Cancer. Biomolecules 2020, 10, 568.
- Gueant, J.L.; Oussalah, A.; Zgheib, R.; Siblini, Y.; Hsu, S.B.; Namour, F. Genetic, epigenetic and genomic mechanisms of methionine dependency of cancer and tumor-initiating cells: What could we learn from folate and methionine cycles. Biochimie 2020, 173, 123–128.
- Zgheib, R.; Battaglia-Hsu, S.F.; Hergalant, S.; Quere, M.; Alberto, J.M.; Chery, C.; Rouyer, P.; Gauchotte, G.; Gueant, J.L.; Namour, F. Folate can promote the methionine-dependent reprogramming of glioblastoma cells towards pluripotency. Cell Death Dis. 2019, 10, 596.
- Wang, Z.; Yip, L.Y.; Lee, J.H.J.; Wu, Z.; Chew, H.Y.; Chong, P.K.W.; Teo, C.C.; Ang, H.Y.; Peh, K.L.E.; Yuan, J.; et al. Methionine is a metabolic dependency of tumor-initiating cells. Nat. Med. 2019, 25, 825–837.
- Strekalova, E.; Malin, D.; Weisenhorn, E.M.M.; Russell, J.D.; Hoelper, D.; Jain, A.; Coon, J.J.; Lewis, P.W.; Cryns, V.L. S-adenosylmethionine biosynthesis is a targetable metabolic vulnerability of cancer stem cells. Breast Cancer Res. Treat 2019, 175, 39–50.
- Altundag, O.; Canpinar, H.; Celebi-Saltik, B. Methionine affects the expression of pluripotency genes and protein levels associated with methionine metabolism in adult, fetal, and cancer stem cells. J. Cell. Biochem. 2022, 123, 406–416.
- Akella, N.M.; Le Minh, G.; Ciraku, L.; Mukherjee, A.; Bacigalupa, Z.A.; Mukhopadhyay, D.; Sodi, V.L.; Reginato, M.J. O-GlcNAc Transferase Regulates Cancer Stem-like Potential of Breast Cancer Cells. Mol. Cancer Res. 2020, 18, 585–598.
- Zhang, W.C.; Shyh-Chang, N.; Yang, H.; Rai, A.; Umashankar, S.; Ma, S.; Soh, B.S.; Sun, L.L.; Tai, B.C.; Nga, M.E.; et al. Glycine decarboxylase activity drives non-small cell lung cancer tumor-initiating cells and tumorigenesis. Cell 2012, 148, 259–272.
- Tian, S.; Feng, J.; Cao, Y.; Shen, S.; Cai, Y.; Yang, D.; Yan, R.; Wang, L.; Zhang, H.; Zhong, X.; et al. Glycine cleavage system determines the fate of pluripotent stem cells via the regulation of senescence and epigenetic modifications. Life Sci. Alliance 2019, 2, e201900413.
- Tsogtbaatar, E.; Landin, C.; Minter-Dykhouse, K.; Folmes, C.D.L. Energy Metabolism Regulates Stem Cell Pluripotency. Front. Cell Dev. Biol. 2020, 8, 87.
- Kang, P.J.; Zheng, J.; Lee, G.; Son, D.; Kim, I.Y.; Song, G.; Park, G.; You, S. Glycine decarboxylase regulates the maintenance and induction of pluripotency via metabolic control. Metab. Eng. 2019, 53, 35–47.
- Chisari, A.; Golan, I.; Campisano, S.; Gelabert, C.; Moustakas, A.; Sancho, P.; Caja, L. Glucose and Amino Acid Metabolic Dependencies Linked to Stemness and Metastasis in Different Aggressive Cancer Types. Front. Pharmacol. 2021, 12, 723798.
- Fuentes-Garcia, G.; Castaneda-Patlan, M.C.; Vercoutter-Edouart, A.S.; Lefebvre, T.; Robles-Flores, M. O-GlcNAcylation Is Involved in the Regulation of Stem Cell Markers Expression in Colon Cancer Cells. Front. Endocrinol. 2019, 10, 289.
- Gueant, J.L.; Namour, F.; Gueant-Rodriguez, R.M.; Daval, J.L. Folate and fetal programming: A play in epigenomics? Trends Endocrinol. Metab. 2013, 24, 279–289.
- Kosgei, V.J.; Coelho, D.; Gueant-Rodriguez, R.M.; Gueant, J.L. Sirt1-PPARS Cross-Talk in Complex Metabolic Diseases and Inherited Disorders of the One Carbon Metabolism. Cells 2020, 9, 1882.
- Gueant, J.L.; Elakoum, R.; Ziegler, O.; Coelho, D.; Feigerlova, E.; Daval, J.L.; Gueant-Rodriguez, R.M. Nutritional models of foetal programming and nutrigenomic and epigenomic dysregulations of fatty acid metabolism in the liver and heart. Pflugers Arch. 2014, 466, 833–850.
- Gueant, J.L.; Gueant-Rodriguez, R.M.; Kosgei, V.J.; Coelho, D. Causes and consequences of impaired methionine synthase activity in acquired and inherited disorders of vitamin B12 metabolism. Crit. Rev. Biochem. Mol. Biol. 2022, 57, 133–155.
- Garcia, M.M.; Gueant-Rodriguez, R.M.; Pooya, S.; Brachet, P.; Alberto, J.M.; Jeannesson, E.; Maskali, F.; Gueguen, N.; Marie, P.Y.; Lacolley, P.; et al. Methyl donor deficiency induces cardiomyopathy through altered methylation/acetylation of PGC-1alpha by PRMT1 and SIRT1. J. Pathol. 2011, 225, 324–335.
- Chen, A.C.H.; Peng, Q.; Fong, S.W.; Lee, K.C.; Yeung, W.S.B.; Lee, Y.L. DNA Damage Response and Cell Cycle Regulation in Pluripotent Stem Cells. Genes 2021, 12, 1548.
- Lee, Y.L.; Peng, Q.; Fong, S.W.; Chen, A.C.; Lee, K.F.; Ng, E.H.; Nagy, A.; Yeung, W.S. Sirtuin 1 facilitates generation of induced pluripotent stem cells from mouse embryonic fibroblasts through the miR-34a and p53 pathways. PLoS ONE 2012, 7, e45633.
- Williams, E.O.; Taylor, A.K.; Bell, E.L.; Lim, R.; Kim, D.M.; Guarente, L. Sirtuin 1 Promotes Deacetylation of Oct4 and Maintenance of Naive Pluripotency. Cell Rep. 2016, 17, 809–820.
- Mu, W.L.; Wang, Y.J.; Xu, P.; Hao, D.L.; Liu, X.Z.; Wang, T.T.; Chen, F.; Chen, H.Z.; Lv, X.; Liu, D.P. Sox2 Deacetylation by Sirt1 Is Involved in Mouse Somatic Reprogramming. Stem Cells 2015, 33, 2135–2147.
- Geoffroy, A.; Saber-Cherif, L.; Pourie, G.; Helle, D.; Umoret, R.; Gueant, J.L.; Bossenmeyer-Pourie, C.; Daval, J.L. Developmental Impairments in a Rat Model of Methyl Donor Deficiency: Effects of a Late Maternal Supplementation with Folic Acid. Int. J. Mol. Sci. 2019, 20, 973.
- Zhang, Z.N.; Chung, S.K.; Xu, Z.; Xu, Y. Oct4 maintains the pluripotency of human embryonic stem cells by inactivating p53 through Sirt1-mediated deacetylation. Stem Cells 2014, 32, 157–165.
- Jang, J.; Huh, Y.J.; Cho, H.J.; Lee, B.; Park, J.; Hwang, D.Y.; Kim, D.W. SIRT1 Enhances the Survival of Human Embryonic Stem Cells by Promoting DNA Repair. Stem Cell Rep. 2017, 9, 629–641.
- De Bonis, M.L.; Ortega, S.; Blasco, M.A. SIRT1 is necessary for proficient telomere elongation and genomic stability of induced pluripotent stem cells. Stem Cell Rep. 2014, 2, 690–706.
- Wang, M.J.; Chen, J.J.; Song, S.H.; Su, J.; Zhao, L.H.; Liu, Q.G.; Yang, T.; Chen, Z.; Liu, C.; Fu, Z.R.; et al. Inhibition of SIRT1 Limits Self-Renewal and Oncogenesis by Inducing Senescence of Liver Cancer Stem Cells. J. Hepatocell Carcinoma 2021, 8, 685–699.
- Shi, L.; Tang, X.; Qian, M.; Liu, Z.; Meng, F.; Fu, L.; Wang, Z.; Zhu, W.G.; Huang, J.D.; Zhou, Z.; et al. A SIRT1-centered circuitry regulates breast cancer stemness and metastasis. Oncogene 2018, 37, 6299–6315.
- Liu, L.; Liu, C.; Zhang, Q.; Shen, J.; Zhang, H.; Shan, J.; Duan, G.; Guo, D.; Chen, X.; Cheng, J.; et al. SIRT1-mediated transcriptional regulation of SOX2 is important for self-renewal of liver cancer stem cells. Hepatology 2016, 64, 814–827.
- Carafa, V.; Altucci, L.; Nebbioso, A. Dual Tumor Suppressor and Tumor Promoter Action of Sirtuins in Determining Malignant Phenotype. Front. Pharmacol. 2019, 10, 38.
- Lim, C.S. SIRT1: Tumor promoter or tumor suppressor? Med. Hypotheses 2006, 67, 341–344.
- Garcia-Peterson, L.M.; Li, X. Trending topics of SIRT1 in tumorigenicity. Biochim. Biophys. Acta Gen. Subj. 2021, 1865, 129952.
- Chen, X.; Sun, K.; Jiao, S.; Cai, N.; Zhao, X.; Zou, H.; Xie, Y.; Wang, Z.; Zhong, M.; Wei, L. High levels of SIRT1 expression enhance tumorigenesis and associate with a poor prognosis of colorectal carcinoma patients. Sci. Rep. 2014, 4, 7481.
- Haynes, H.R.; Scott, H.L.; Killick-Cole, C.L.; Shaw, G.; Brend, T.; Hares, K.M.; Redondo, J.; Kemp, K.C.; Ballesteros, L.S.; Herman, A.; et al. shRNA-mediated PPARalpha knockdown in human glioma stem cells reduces in vitro proliferation and inhibits orthotopic xenograft tumour growth. J. Pathol. 2019, 247, 422–434.
- Lee, J.; Lee, J.; Cho, Y.S. Peroxisome Proliferator-Activated Receptor alpha Agonist and Its Target Nanog Cooperate to Induce Pluripotency. J. Clin. Med. 2018, 7, 488.
- Papi, A.; Storci, G.; Guarnieri, T.; De Carolis, S.; Bertoni, S.; Avenia, N.; Sanguinetti, A.; Sidoni, A.; Santini, D.; Ceccarelli, C.; et al. Peroxisome proliferator activated receptor-alpha/hypoxia inducible factor-1alpha interplay sustains carbonic anhydrase IX and apoliprotein E expression in breast cancer stem cells. PLoS ONE 2013, 8, e54968.
- Papi, A.; Guarnieri, T.; Storci, G.; Santini, D.; Ceccarelli, C.; Taffurelli, M.; De Carolis, S.; Avenia, N.; Sanguinetti, A.; Sidoni, A.; et al. Nuclear receptors agonists exert opposing effects on the inflammation dependent survival of breast cancer stem cells. Cell Death Differ. 2012, 19, 1208–1219.


