1. Methionine in Normal Stem Cells
The work of Shiraki et al. on human embryonic stem cells (hESCs) and human induced pluripotent stem cells (hiPSCs) highlighted the high-Met metabolic state of these cells compared to differentiated cells and their dependence on Met in a concentration-dependent manner. Their work was inspired by the specialized metabolic state of stem cells and the dependence of mouse ESCs on threonine catabolism [
27]. Mouse embryonic stem cells demonstrate a high-flux metabolic state with a high expression of threonine dehydrogenase (TDH). The TDH enzyme catalyses a rate-limiting step in the mitochondrial conversion of threonine into glycine and acetyl-coenzyme A (CoA), which are essential for the folate cycle and tricarboxylic acid (TCA) cycle, respectively. This makes mouse ESCs critically dependent on threonine for their survival, pluripotency maintenance, and differentiation [
27]. Given that threonine dehydrogenase is a non-functional pseudogene in humans, Shiraki et al. sought an equivalent approach to detect whether a similar effect could be observed with other amino acids in human cells by depriving them of single amino acids and measuring the total number of cells 48 h after deprivation. The most striking results were observed in the Met-deprived condition [
12]. Furthermore, they showed that short Met deprivation led to a fast decrease in intracellular SAM with a consequent decrease in H3K4me3, and a decrease in NANOG expression, along with an increase in the differentiation potency in the three germ layers [
12]. It is also interesting to note that the supplementation of Met increased NANOG expression and decreased the proportion of p53+ cells in a concentration-dependent manner [
12]. In contrast, the prolonged Met deprivation effect on the cells was irreversible, with G0/G1 phase arrest triggering apoptosis. This leads to the conclusion that hESCs/hiPSCs do not rely on threonine-derived one-carbon metabolism for self-renewal and pluripotency, and alternatively use Met metabolism to achieve the same result [
11,
12]. Recently, the same team showed the involvement of zinc signaling in the regulation of stem cell pluripotency and differentiation. Culturing PSCs in Zn-deprived medium partially mimicked methionine deprivation (e.g., potentiated differentiation), showing altered methionine metabolism-related metabolite profiles. Likewise, the depletion of methionine reduced protein-bound Zn, which includes MS, through displacement with homocysteine [
28].
The increase in Met requirements in the stemness and self-renewal capacity of ESCs/iPSCs is related to SAM levels and the synthesis of SAM by MAT2A and MAT2B. Knocking down MAT2A or MAT2B hampers the cell’s ability to transform Met into SAM and leads to a decreased self-renewal capacity that can be rescued with SAM addition. The Met salvage pathway seems to play a limited role in the adaption of ESCs/iPSCs cells to their increased Met needs. The knocking down of SMS, the enzyme synthesizing spermine, impairs the Met salvage pathway, but does not phenocopy the Met depletion condition [
12]. In addition, cycloleucine, an analog of Met that specifically inhibits MAT, decreases SAM levels and cell growth without affecting Met or SAH levels. Met restriction induces the upregulation of MAT2A expression as a way for the cells to possibly increase SAM levels and cope with the restrictive conditions [
12]. These results indicate that SAM, rather than Met itself, is essential for the self-renewal and survival of stem cells. However, in cells with knocked down SIRT1, MAT2A is downregulated, and the upregulation induced through Met restriction leads to a limited increase in MAT2A level, compared to the wild-type (WT) condition. This downregulation is triggered by c-Myc, a proliferation proto-oncogene transregulator under the control of SIRT1 through a deacetylation mechanism that increases its stability and activity [
10].
Adenosyl homocysteinase (AHCY) is another target enzyme of the Met cycle, which may influence the relation between SAM and cell stemness. AHCY catalyzes the hydrolysis of S-adenosylhomocysteine (SAH) to generate adenine and homocysteine as a part of the Met cycle [
8]. Inducing the differentiation of mESCs decreases the mRNA levels of AHCY, MAT2A, MAT2B SAM and SAH, and homocysteine metabolites of the Met cycle. Depleting AHCY using shRNA leads to a decrease in the SAM/SAH ratio and pluripotency markers, notably Oct4 and Nanog, and an increase in the differentiation markers. Furthermore, the depletion results in an increase in cell number in the G1 phase, accompanied by a reduction in cells in the S phase and activation of the p53-dependent signaling pathway, leading to increased apoptosis [
8]. The decrease in the SAM/SAH ratio leads to a decrease in H3K4me3 levels, notably at the Pou5f1 and Nanog loci and the O-GlcNAc post-translational modifications on serine or threonine residues of nucleocytoplasmic proteins [
8]. O-GlcNAcylation is a nutrient-responsive modification with a pivotal role in stem cell biology [
29,
30,
31]. This post-translational modification modulates enzyme activities. AHCY undergoes T136 O-GlcNAcylation, which promotes its activity by increasing its tetrameric assembly and its affinity with Hcy [
8]. Inducing mESC differentiation leads to a gradual decrease in AHCY O-GlcNAcylation with reduced enzyme activity, thus regulating mESC pluripotency and self-renewal capacity [
8] (
Figure 2).
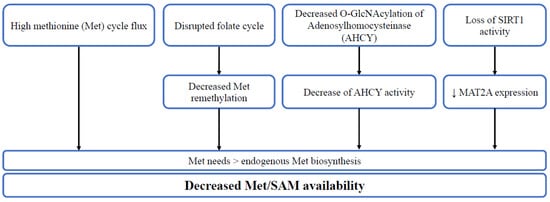
Figure 2. The diverse mechanisms leading to decreased Met/SAM availability. High methionine cycle flux, disrupted folate cycle, decreased O-GlcNAcylation of adenosylhomocysteinase (AHCY) or loss of SIRT1 activity lead to higher methionine needs in comparison to endogenous synthesis, thus decreasing methionine/SAM availability.
2. Methionine in Cancer Stem Cells
The growth of many cancer cells depends on Met cellular availability [
32,
33,
34]. The Met dependency of cancer cells is defined by the inability of cells to proliferate in a medium deprived of Met, even when the metabolic precursor of Met, Hcy, is present [
32,
33,
34]. Recent studies have shown that cancer stem cells are Met-dependent. The disruption of one-carbon metabolism enzymes or the reduction in Met hamper the self-renewal capacity and pluripotency of cancer stem cells [
3,
4,
5,
7,
9].
Zgheib et al. found evidence of Met dependency in glioblastoma cancer stem cells, but not in the related adherent differentiated cells. The cells’ tumorsphere formation capacities are recovered upon adding folate or MeTHF major molecules in the folate cycle. Glioblastoma stem cells demonstrated a disrupted folate cycle and could not furnish the necessary one-carbon unit to convert it into Met and SAM [
3]. In contrast, the Met dependency stems from the high need for SAM in lung TICs [
4]. Lung TICs have a high Met cycle flux, leading to their dependency on exogenous Met, as observed for normal stem cells. Compared to their differentiated counterpart, these cells demonstrate high GLDC expression and activity [
4]. The high activity of GLDC in TICs redirects the flux of one-carbon units towards the increased demand of the Met cycle [
4,
35]. Knocking down this gene decreases Met and SAH levels to a level equivalent to differentiated cells, decreases histone methylation, and hampers the tumor-initiating capacities of these cells, further demonstrating the importance of Met [
4]. Interestingly, recent studies have shown the importance of GLDC in maintaining and inducing pluripotency in ESCs and iPSCs through fueling H3K4me3 modification, while knocking down GLDC suppressed the pluripotency state [
26,
36,
37]. Transient (48 h) Met starvation of lung TICs led to more striking results, with a decrease in SAM, SAH, and histone methylation levels and again hampered tumor-initiating capacities [
4]. Supplementing cells with Hcy does not allow the cells to recover from the effect of Met starvation, in agreement with the Met dependency definition described above. Adding SAM or replating cells in Met-supplemented media allowed the cells to recover and regain their tumor-initiating ability [
4]. Moreover, three other enzymes, MAT2A, MTHFR, and SHMT2, are also elevated in TICs compared to differentiated cells, further confirming the impact of GLDC upregulation of TICs on Met cycle flux. Furthermore, in cases where any of these genes are knocked down, the cell’s ability to form tumors is hampered, similarly knocking down GLDC [
4]. Both MAT2A and MTHFR were found to be upregulated in lung tumors, but only MAT2A was found to be upregulated in high-grade primary tumors or metastasis, highlighting the possibility of using MAT2A inhibitors, such as FIDAS-5, to treat patients with aggressive lung tumors. Wang et al. found that it hampers the tumor-initiating ability of TICs [
4]. The mechanisms of Met dependency observed in TICs from glioblastoma and lung cancer cells is, thus, due to the disruption of the balance between folate and methionine cycles, leading to excess or impaired production of methyl-THF and methionine, which limits their growth [
34]. Overall, these results suggest a prominent role of one-carbon metabolism in conferring stemness in cancer cells [
3,
34,
38].
The dependence on Met in glioblastoma TICs seems to have a bell shape depending on the concentration used, with an optimal concentration. Therefore, lower or higher concentrations lead to lower tumorsphere formation capacity [
3]. Such a correlation was not investigated in normal stem cells, as the limit of the tested concentration of Met was approximately 120 µM in comparison to an upper concentration of 500 µM in the study on glioblastoma TICs [
3,
7,
12]. In normal stem cells, the concentration of Met was positively correlated with the increase in pluripotency gene expression and lower mortality rates were observed with a higher concentration of Met [
12]. Cultivating breast cancer stem cells in Met-deprived conditions mirrors the results obtained in normal stem cells cultured in similar conditions [
5,
9]. The mammosphere formation assay shows the significantly lower capacity of cancer stem cells to form in those conditions, possibly through the same mechanism observed in ESCs/iPSCs with a recovered capacity upon adding SAM [
5,
9]. In addition, the Met-deprived condition leads to the increase in MAT2A levels as a homeostatic response and a decrease in H3K4me3 levels with lower levels of SOX9 markers and CD44+/CD24−. Intriguingly, these conditions potentiate the knocking down effect of MAT2A or its inhibition by MAT2A inhibitors. Similarly, mice fed a Met-restricted diet and treated with MAT2A inhibitor show a potentiated result, with increased apoptosis, lower tumor volume, and metastatic capacity compared to the control conditions [
5]. Breast cancer stem cells were also found to highly express the O-GlcNAc transferase enzyme (OGT) and O-GlcNAcylation [
9]. In line with what was found in normal stem cells, altering OGT and O-GlcNAc levels hampers stem cells’ ability to form mammospheres in vitro, tumor formation in vivo, and lowered the levels of CD44+CD24− [
9]. In addition, OGT seems to be a regulator of epithelial–mesenchymal transition (EMT) and cancer stem-like cell markers, including CD44, NANOG, and c-Myc [
9]. Likewise, O-GlcNAcylation is involved in regulating stem cell marker expression in colon cancer cells [
39]. Despite the resemblance in the importance of the O-GlcNAc effect on the stemness between normal and cancer stem cells, they are not linked to AHCY. Thus, the influence of O-GlcNAc on the Met cycle in cancer stem cells needs to be assessed in future studies [
9].
3. The Influence of SIRT1 and PPAR-Alpha/PGC1-Alpha Pathway
SIRT1 and PPAR-α are key players in the links between Met metabolism and cell stemness. SIRT1 is one of the seven mammalian proteins that belong to the sirtuin family [
14,
40,
41]. It catalyzes histone and non-histone lysine deacetylation in a NAD+-dependent manner [
14,
40,
41]. Previous work in our lab showed the role of SIRT1 in regulating energy metabolism through PPAR-α and PGC-1α in a methyl-deficient diet [
14,
40,
41,
42,
43]. PGC-1α is a master regulator of lipid metabolism and fatty acid oxidation. It is regulated by methylation and acetylation. The deacetylation of the PGC-1α protein leads to its activation and is known to coactivate PPAR-α to enhance the expression of fatty acid oxidation genes, antioxidant enzymes, and mitochondrial biogenesis [
40]. Methyl-deficient diets decrease the expression of SIRT1 and subsequent activation of PGC-1α through imbalanced acetylation and methylation of the latter dysregulating energy metabolism [
14,
40,
41]. The impaired expression and/or activity of methionine synthase in fibroblasts from patients with mutations in MTR and/or other inherited disorders of vitamin B12 metabolism also result in decreased protein expression of SIRT1, which plays a key role in the underlying pathological mechanisms of these disorders [
40].
Sirtuin 1 (SIRT1) is involved in iPSC formation [
44]. SIRT1 knockdown decreased, while resveratrol (RSV) increased the efficiency of iPSCs. SIRT1 enhances iPSC generation through deacetylation of p53, inhibition of p21 and enhancement of Nanog expression [
45]. SIRT1 has been demonstrated to interact and acetylate Oct4 to maintain the stemness of naive pluripotent stem cells [
46]. Sirt1 also deacetylates Sox2 through direct interaction with Oct4 [
47]. The B12 and folate deficiency decreases the endogenous synthesis of methionine and decreases the brain expression of miR-34a in pups from deficient mother rats [
48]. Of note, miR-34a reduces the reprogramming efficiency through inhibition of SIRT1 expression [
45]. Decreased SIRT1 leads to the stabilization and increased activity of the P53 protein through its increased acetylation at the K120 and K164 sites [
49]. This SIRT1-dependent upregulation of P53 activity is effective in undifferentiated hESCs, but not in other cell types. SIRT1 plays a role in DNA damage repair that is crucial for hESCs’ fast mitotic division, which is prone to replication-related DNA errors [
50]. It leads to programmed cell death through decreased expression of DNA repair enzymes, such as MSH2, MSH6 and APEX1, in hESCs [
50]. Furthermore, SIRT1 is essential for telomere elongation during the iPSC generation process [
51]. The level of expression of SIRT1 has been demonstrated to be elevated in normal and cancer stem cells (CSC), compared to their differentiated counterparts [
45,
46,
50,
51,
52,
53,
54]. However, there is a debate on the role of SIRT1 in CSCs with its double functionality as a tumor suppressor and promoter [
55,
56]. SIRT 1 expression is increased, and its activity is critical for stemness and cell survival in cancer stem cells from glioma, colon and liver cancer, and leukemia [
57]. SIRT1 inhibits DNMT3A and promotes the expression of SOX2 through promoter-reduced methylation [
54]. It also increases the expression of other stemness-associated genes, including Oct4, Nanog, Cripto, Tert and Lin28, in colon cancer stem cells [
58].
Several studies have investigated the influence of Met restriction on the effects of SIRT1 on pluripotency. However, whether SIRT1 influences stemness through its decreased expression produced by the impaired remethylation pathway of Met metabolism is unknown. Kilberg et al. showed that SIRT1 knock-out produces effects similar to those caused by Met restriction observed by Shiraki et al., in mouse embryonic stem cells (mESCs). The SIRT1 knock-out (KO) mESCs specifically impaired Met metabolism [
10]. The metabolomic analysis of the SIRT1 KO cells cultured in complete media showed an elevated Met level, along with a decrease in SAM levels. The restriction of Met led to elevated differentiation markers and induced apoptosis with high sensitivity to Met deprivation, compared to other amino acids [
10]. Interestingly, culturing SIRT1 KO mESCs in a complete medium with normal levels of Met demonstrated SAM levels similar to those of WT mESCs after Met restriction, suggesting that knocking out SIRT1 is somewhat equivalent to Met restriction, even with normal Met levels in culture medium [
10]. Furthermore, SIRT1 KO reduced NANOG and OCT4 expression, with a marked decrease in NANOG expression when cultured in Met-restricted medium. In line with these results, the relative levels of H3K4me3 at the transcription starting site of the Nanog gene were reduced by both SIRT1 deletion and Met restriction, possibly through the same mechanism observed in hESCs/hiPSCs and steered by SAM levels [
10].
PPAR-α is a key player in stemness. It triggers the expression of key genes of pluripotency reprogramming and enables pluripotent cells to adapt to their metabolic needs [
59,
60,
61,
62]. A Food and Drug Administration (FDA)-approved PPAR-α agonist was found to facilitate iPSC generation and enhance their programming efficiency by increasing the expression of pluripotency genes, including Nanog, Nr5A2, Oct4, and Rex1 [
60]. PPAR-α knockdown of human glioma stem cells by shRNA reduces in vitro proliferation and inhibits orthotopic xenograft tumor growth [
59]. Furthermore, PPAR-alpha was shown to play an important role in promoting mammosphere formation by modulating the expression of stem cell genes, including Jagged1, via the NF-κB/IL6 axis [
62]. The PPARα-specific agonist treatment increases the number of mammospheres [
62]. On the other hand, PPARα siRNA conditions decrease the number of mammospheres [
62]. Whether the decreased activity of PPAR-α is related to the inactivation of PGC-1α by SIRT1 in stem cells has not been considered in experimental studies. However, this hypothesis
This entry is adapted from the peer-reviewed paper 10.3390/cells11223607