Stem cells are a population of undifferentiated cells with self-renewal and differentiation capacities. Normal and cancer stem cells share similar characteristics in relation to their stemness properties. One-carbon metabolism (OCM), a network of interconnected reactions, plays an important role in this dependence through its role in the endogenous synthesis of methionine and S-adenosylmethionine (SAM), the universal donor of methyl groups in eukaryotic cells. OCM genes are differentially expressed in stem cells, compared to their differentiated counterparts. Furthermore, cultivating stem cells in methionine-restricted conditions hinders their stemness capacities through decreased SAM levels with a subsequent decrease in histone methylation, notably H3K4me3, with a decrease in stem cell markers. Stem cells’ reliance on methionine is linked to several mechanisms, including high methionine flux or low endogenous methionine biosynthesis.
- normal stem cells
- cancer stem cells
- methionine
- methionine dependence
- stemness
1. Methionine in Normal Stem Cells
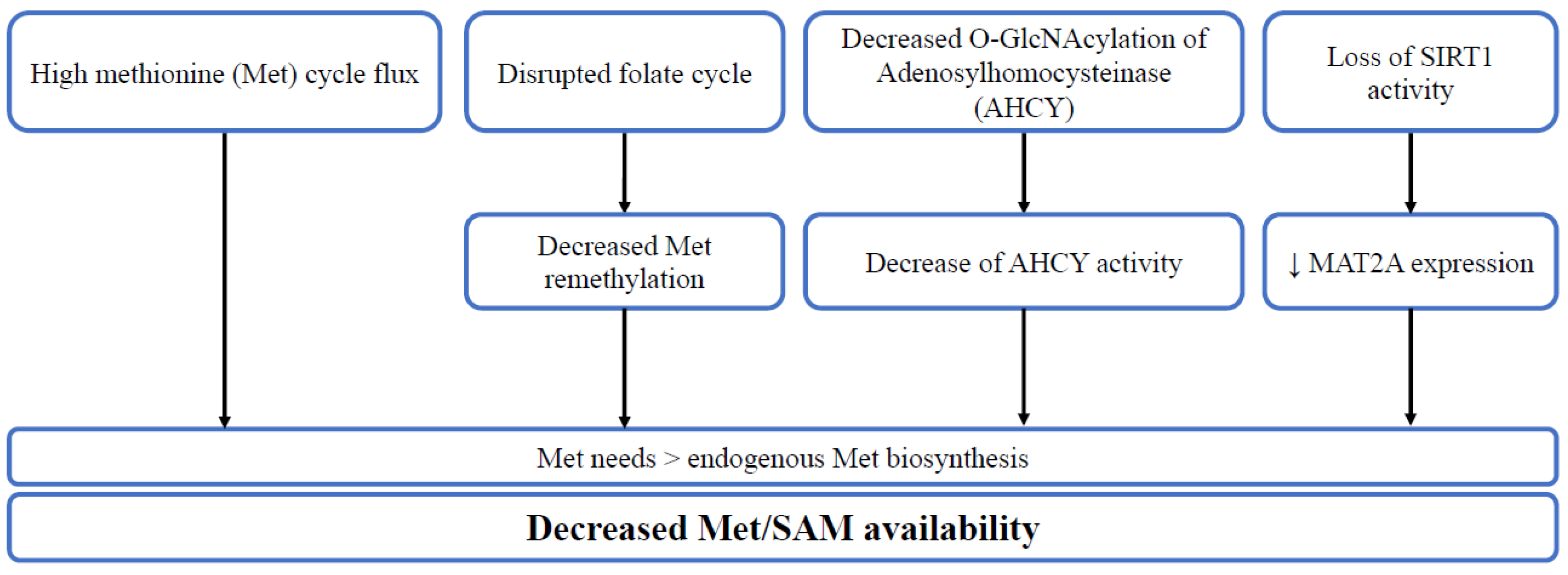
2. Methionine in Cancer Stem Cells
3. The Influence of SIRT1 and PPAR-Alpha/PGC1-Alpha Pathway
References
- Wang, J.; Alexander, P.; Wu, L.; Hammer, R.; Cleaver, O.; McKnight, S.L. Dependence of mouse embryonic stem cells on threonine catabolism. Science 2009, 325, 435–439.
- Shiraki, N.; Shiraki, Y.; Tsuyama, T.; Obata, F.; Miura, M.; Nagae, G.; Aburatani, H.; Kume, K.; Endo, F.; Kume, S. Methionine metabolism regulates maintenance and differentiation of human pluripotent stem cells. Cell Metab. 2014, 19, 780–794.
- Kilberg, M.S.; Terada, N.; Shan, J. Influence of Amino Acid Metabolism on Embryonic Stem Cell Function and Differentiation. Adv. Nutr. 2016, 7, 780S–789S.
- Sim, E.Z.; Enomoto, T.; Shiraki, N.; Furuta, N.; Kashio, S.; Kambe, T.; Tsuyama, T.; Arakawa, A.; Ozawa, H.; Yokoyama, M.; et al. Methionine metabolism regulates pluripotent stem cell pluripotency and differentiation through zinc mobilization. Cell Rep. 2022, 40, 111120.
- Tang, S.; Fang, Y.; Huang, G.; Xu, X.; Padilla-Banks, E.; Fan, W.; Xu, Q.; Sanderson, S.M.; Foley, J.F.; Dowdy, S.; et al. Methionine metabolism is essential for SIRT1-regulated mouse embryonic stem cell maintenance and embryonic development. EMBO J. 2017, 36, 3175–3193.
- Zhu, Q.; Cheng, X.; Cheng, Y.; Chen, J.; Xu, H.; Gao, Y.; Duan, X.; Ji, J.; Li, X.; Yi, W. O-GlcNAcylation regulates the methionine cycle to promote pluripotency of stem cells. Proc. Natl. Acad. Sci. USA 2020, 117, 7755–7763.
- Hanover, J.A.; Krause, M.W.; Love, D.C. Bittersweet memories: Linking metabolism to epigenetics through O-GlcNAcylation. Nat. Rev. Mol. Cell Biol. 2012, 13, 312–321.
- Sheikh, M.A.; Emerald, B.S.; Ansari, S.A. Stem cell fate determination through protein O-GlcNAcylation. J. Biol. Chem. 2021, 296, 100035.
- Wells, L.; Vosseller, K.; Hart, G.W. Glycosylation of nucleocytoplasmic proteins: Signal transduction and O-GlcNAc. Science 2001, 291, 2376–2378.
- Sorin, M.; Watkins, D.; Gilfix, B.M.; Rosenblatt, D.S. Methionine dependence in tumor cells: The potential role of cobalamin and MMACHC. Mol. Genet. Metab. 2021, 132, 155–161.
- Kaiser, P. Methionine Dependence of Cancer. Biomolecules 2020, 10, 568.
- Gueant, J.L.; Oussalah, A.; Zgheib, R.; Siblini, Y.; Hsu, S.B.; Namour, F. Genetic, epigenetic and genomic mechanisms of methionine dependency of cancer and tumor-initiating cells: What could we learn from folate and methionine cycles. Biochimie 2020, 173, 123–128.
- Zgheib, R.; Battaglia-Hsu, S.F.; Hergalant, S.; Quere, M.; Alberto, J.M.; Chery, C.; Rouyer, P.; Gauchotte, G.; Gueant, J.L.; Namour, F. Folate can promote the methionine-dependent reprogramming of glioblastoma cells towards pluripotency. Cell Death Dis. 2019, 10, 596.
- Wang, Z.; Yip, L.Y.; Lee, J.H.J.; Wu, Z.; Chew, H.Y.; Chong, P.K.W.; Teo, C.C.; Ang, H.Y.; Peh, K.L.E.; Yuan, J.; et al. Methionine is a metabolic dependency of tumor-initiating cells. Nat. Med. 2019, 25, 825–837.
- Strekalova, E.; Malin, D.; Weisenhorn, E.M.M.; Russell, J.D.; Hoelper, D.; Jain, A.; Coon, J.J.; Lewis, P.W.; Cryns, V.L. S-adenosylmethionine biosynthesis is a targetable metabolic vulnerability of cancer stem cells. Breast Cancer Res. Treat 2019, 175, 39–50.
- Altundag, O.; Canpinar, H.; Celebi-Saltik, B. Methionine affects the expression of pluripotency genes and protein levels associated with methionine metabolism in adult, fetal, and cancer stem cells. J. Cell. Biochem. 2022, 123, 406–416.
- Akella, N.M.; Le Minh, G.; Ciraku, L.; Mukherjee, A.; Bacigalupa, Z.A.; Mukhopadhyay, D.; Sodi, V.L.; Reginato, M.J. O-GlcNAc Transferase Regulates Cancer Stem-like Potential of Breast Cancer Cells. Mol. Cancer Res. 2020, 18, 585–598.
- Zhang, W.C.; Shyh-Chang, N.; Yang, H.; Rai, A.; Umashankar, S.; Ma, S.; Soh, B.S.; Sun, L.L.; Tai, B.C.; Nga, M.E.; et al. Glycine decarboxylase activity drives non-small cell lung cancer tumor-initiating cells and tumorigenesis. Cell 2012, 148, 259–272.
- Tian, S.; Feng, J.; Cao, Y.; Shen, S.; Cai, Y.; Yang, D.; Yan, R.; Wang, L.; Zhang, H.; Zhong, X.; et al. Glycine cleavage system determines the fate of pluripotent stem cells via the regulation of senescence and epigenetic modifications. Life Sci. Alliance 2019, 2, e201900413.
- Tsogtbaatar, E.; Landin, C.; Minter-Dykhouse, K.; Folmes, C.D.L. Energy Metabolism Regulates Stem Cell Pluripotency. Front. Cell Dev. Biol. 2020, 8, 87.
- Kang, P.J.; Zheng, J.; Lee, G.; Son, D.; Kim, I.Y.; Song, G.; Park, G.; You, S. Glycine decarboxylase regulates the maintenance and induction of pluripotency via metabolic control. Metab. Eng. 2019, 53, 35–47.
- Chisari, A.; Golan, I.; Campisano, S.; Gelabert, C.; Moustakas, A.; Sancho, P.; Caja, L. Glucose and Amino Acid Metabolic Dependencies Linked to Stemness and Metastasis in Different Aggressive Cancer Types. Front. Pharmacol. 2021, 12, 723798.
- Fuentes-Garcia, G.; Castaneda-Patlan, M.C.; Vercoutter-Edouart, A.S.; Lefebvre, T.; Robles-Flores, M. O-GlcNAcylation Is Involved in the Regulation of Stem Cell Markers Expression in Colon Cancer Cells. Front. Endocrinol. 2019, 10, 289.
- Gueant, J.L.; Namour, F.; Gueant-Rodriguez, R.M.; Daval, J.L. Folate and fetal programming: A play in epigenomics? Trends Endocrinol. Metab. 2013, 24, 279–289.
- Kosgei, V.J.; Coelho, D.; Gueant-Rodriguez, R.M.; Gueant, J.L. Sirt1-PPARS Cross-Talk in Complex Metabolic Diseases and Inherited Disorders of the One Carbon Metabolism. Cells 2020, 9, 1882.
- Gueant, J.L.; Elakoum, R.; Ziegler, O.; Coelho, D.; Feigerlova, E.; Daval, J.L.; Gueant-Rodriguez, R.M. Nutritional models of foetal programming and nutrigenomic and epigenomic dysregulations of fatty acid metabolism in the liver and heart. Pflugers Arch. 2014, 466, 833–850.
- Gueant, J.L.; Gueant-Rodriguez, R.M.; Kosgei, V.J.; Coelho, D. Causes and consequences of impaired methionine synthase activity in acquired and inherited disorders of vitamin B12 metabolism. Crit. Rev. Biochem. Mol. Biol. 2022, 57, 133–155.
- Garcia, M.M.; Gueant-Rodriguez, R.M.; Pooya, S.; Brachet, P.; Alberto, J.M.; Jeannesson, E.; Maskali, F.; Gueguen, N.; Marie, P.Y.; Lacolley, P.; et al. Methyl donor deficiency induces cardiomyopathy through altered methylation/acetylation of PGC-1alpha by PRMT1 and SIRT1. J. Pathol. 2011, 225, 324–335.
- Chen, A.C.H.; Peng, Q.; Fong, S.W.; Lee, K.C.; Yeung, W.S.B.; Lee, Y.L. DNA Damage Response and Cell Cycle Regulation in Pluripotent Stem Cells. Genes 2021, 12, 1548.
- Lee, Y.L.; Peng, Q.; Fong, S.W.; Chen, A.C.; Lee, K.F.; Ng, E.H.; Nagy, A.; Yeung, W.S. Sirtuin 1 facilitates generation of induced pluripotent stem cells from mouse embryonic fibroblasts through the miR-34a and p53 pathways. PLoS ONE 2012, 7, e45633.
- Williams, E.O.; Taylor, A.K.; Bell, E.L.; Lim, R.; Kim, D.M.; Guarente, L. Sirtuin 1 Promotes Deacetylation of Oct4 and Maintenance of Naive Pluripotency. Cell Rep. 2016, 17, 809–820.
- Mu, W.L.; Wang, Y.J.; Xu, P.; Hao, D.L.; Liu, X.Z.; Wang, T.T.; Chen, F.; Chen, H.Z.; Lv, X.; Liu, D.P. Sox2 Deacetylation by Sirt1 Is Involved in Mouse Somatic Reprogramming. Stem Cells 2015, 33, 2135–2147.
- Geoffroy, A.; Saber-Cherif, L.; Pourie, G.; Helle, D.; Umoret, R.; Gueant, J.L.; Bossenmeyer-Pourie, C.; Daval, J.L. Developmental Impairments in a Rat Model of Methyl Donor Deficiency: Effects of a Late Maternal Supplementation with Folic Acid. Int. J. Mol. Sci. 2019, 20, 973.
- Zhang, Z.N.; Chung, S.K.; Xu, Z.; Xu, Y. Oct4 maintains the pluripotency of human embryonic stem cells by inactivating p53 through Sirt1-mediated deacetylation. Stem Cells 2014, 32, 157–165.
- Jang, J.; Huh, Y.J.; Cho, H.J.; Lee, B.; Park, J.; Hwang, D.Y.; Kim, D.W. SIRT1 Enhances the Survival of Human Embryonic Stem Cells by Promoting DNA Repair. Stem Cell Rep. 2017, 9, 629–641.
- De Bonis, M.L.; Ortega, S.; Blasco, M.A. SIRT1 is necessary for proficient telomere elongation and genomic stability of induced pluripotent stem cells. Stem Cell Rep. 2014, 2, 690–706.
- Wang, M.J.; Chen, J.J.; Song, S.H.; Su, J.; Zhao, L.H.; Liu, Q.G.; Yang, T.; Chen, Z.; Liu, C.; Fu, Z.R.; et al. Inhibition of SIRT1 Limits Self-Renewal and Oncogenesis by Inducing Senescence of Liver Cancer Stem Cells. J. Hepatocell Carcinoma 2021, 8, 685–699.
- Shi, L.; Tang, X.; Qian, M.; Liu, Z.; Meng, F.; Fu, L.; Wang, Z.; Zhu, W.G.; Huang, J.D.; Zhou, Z.; et al. A SIRT1-centered circuitry regulates breast cancer stemness and metastasis. Oncogene 2018, 37, 6299–6315.
- Liu, L.; Liu, C.; Zhang, Q.; Shen, J.; Zhang, H.; Shan, J.; Duan, G.; Guo, D.; Chen, X.; Cheng, J.; et al. SIRT1-mediated transcriptional regulation of SOX2 is important for self-renewal of liver cancer stem cells. Hepatology 2016, 64, 814–827.
- Carafa, V.; Altucci, L.; Nebbioso, A. Dual Tumor Suppressor and Tumor Promoter Action of Sirtuins in Determining Malignant Phenotype. Front. Pharmacol. 2019, 10, 38.
- Lim, C.S. SIRT1: Tumor promoter or tumor suppressor? Med. Hypotheses 2006, 67, 341–344.
- Garcia-Peterson, L.M.; Li, X. Trending topics of SIRT1 in tumorigenicity. Biochim. Biophys. Acta Gen. Subj. 2021, 1865, 129952.
- Chen, X.; Sun, K.; Jiao, S.; Cai, N.; Zhao, X.; Zou, H.; Xie, Y.; Wang, Z.; Zhong, M.; Wei, L. High levels of SIRT1 expression enhance tumorigenesis and associate with a poor prognosis of colorectal carcinoma patients. Sci. Rep. 2014, 4, 7481.
- Haynes, H.R.; Scott, H.L.; Killick-Cole, C.L.; Shaw, G.; Brend, T.; Hares, K.M.; Redondo, J.; Kemp, K.C.; Ballesteros, L.S.; Herman, A.; et al. shRNA-mediated PPARalpha knockdown in human glioma stem cells reduces in vitro proliferation and inhibits orthotopic xenograft tumour growth. J. Pathol. 2019, 247, 422–434.
- Lee, J.; Lee, J.; Cho, Y.S. Peroxisome Proliferator-Activated Receptor alpha Agonist and Its Target Nanog Cooperate to Induce Pluripotency. J. Clin. Med. 2018, 7, 488.
- Papi, A.; Storci, G.; Guarnieri, T.; De Carolis, S.; Bertoni, S.; Avenia, N.; Sanguinetti, A.; Sidoni, A.; Santini, D.; Ceccarelli, C.; et al. Peroxisome proliferator activated receptor-alpha/hypoxia inducible factor-1alpha interplay sustains carbonic anhydrase IX and apoliprotein E expression in breast cancer stem cells. PLoS ONE 2013, 8, e54968.
- Papi, A.; Guarnieri, T.; Storci, G.; Santini, D.; Ceccarelli, C.; Taffurelli, M.; De Carolis, S.; Avenia, N.; Sanguinetti, A.; Sidoni, A.; et al. Nuclear receptors agonists exert opposing effects on the inflammation dependent survival of breast cancer stem cells. Cell Death Differ. 2012, 19, 1208–1219.