2. Neurodegenerative Proteinopathies
Neurodegenerative proteinopathies is an umbrella term for neurodegenerative disorders characterized by the formation of misfolded protein aggregates that cause cellular toxicity and contribute to cellular proteostatic collapse
[12]. According to the pathophysiological hypothesis of neurodegenerative disorders, some proteins change their conformations, consequently gaining neurotoxic activity or losing their normal function by forming small oligomeric or large fibrillar aggregates, leading to neurodegeneration
[3]. Neurodegenerative proteinopathies include some of the most common neurodegenerative disorders, such as Alzheimer’s and Parkinson’s disease, as well as Huntington’s disease, multiple sclerosis, amyotrophic lateral sclerosis, etc. The recent findings showing that bassoon proteinopathy drives neurodegeneration in mice and patients with multiple sclerosis are reminiscent of disease pathways in neurodegenerative proteinopathies
[13].
The neuronal proteome consists of 10,000 to 20,000 different proteins that, in order to fulfill their biological function, must fold in accordance with the instructions encoded in the amino acid sequence
[14]. Therefore, to maintain cellular integrity and health, the process of protein folding and its degradation must be well-regulated
[15]. However, their biologically active conformation (the native state) is often marginally stable under normal physiological conditions, and even a small polypeptide of ~100 amino acids can adopt many conformations (~10
30) under different conditions
[14]. It is, therefore, hardly surprising that the process of protein folding is error-prone, leading to misfolded states and off-pathway aggregates
[14]. Due to this susceptibility, cells face a continuous stream of misfolded and aggregated proteins (
Table 1) and require supportive molecular chaperones called heat shock proteins (Hsp) to refold, degrade, and eliminate them to maintain proteome homeostasis
[12].
Table 1. Misfolded and aggregated proteins in neurodegenerative proteinopathies.
Under proteotoxic stress conditions induced by reactive oxygen species (ROS), toxins, cell aging, or disease-related gene mutations, proteins can change conformation. When such misfolded proteins escape cellular quality control, they can begin to aggregate into non-native structures, ranging from oligomers and amorphous assemblies to highly ordered amyloid fibrils and plaques
[16]. These structures have the potential to disrupt proteostasis and thus impair normal cellular function
[15]. Cellular protein homeostasis or proteostasis refers to the integrated activity of cellular mechanisms involved in the regulation of protein synthesis, folding, translocation, assembly/disassembly, and degradation
[17]. For example, the heat shock response and the response to unfolded protein involve the transcriptional regulation of various chaperones (e.g., Hsp70 and Hsp90) and non-chaperone proteins such as transcriptional factors, regulators of the cell cycle, as well as signaling receptors and proteins
[17]. In addition, during the ageing process or in disorders associated with misfolded proteins, cells can undergo proteostatic collapse or a condition associated with the accumulation of ubiquitinated inclusion bodies
[18]. These ubiquitinated inclusion bodies are seen in many neurodegenerative disorders and can directly inhibit or clog the proteasomes
[19].
Notably, only single-chain polypeptides can be degraded by proteasomes, requiring the proteins to be partially or fully unfolded
[20]. Higher-order amyloid aggregates are particularly resistant to degradation and are extremely thermodynamically stable
[21]. This stability contributes to the ability of protein aggregates to propagate in a prion-like manner by changing the normally folded counterparts into pathogenic conformations
[21]. Moreover, after the injection of protein aggregates into the brains of normal animals, they can spread to surrounding neurons and neighboring glial cells and induce a new pathology
[22]. In addition, misfolding of one protein can cause other susceptible proteins to misfold
[22], and therefore aggregates of different misfolded proteins can even be observed in the same patient
[23]. Specifically, a particular type of accumulated misfolded proteins can trigger the misfolding of other unrelated proteins that would be properly folded under normal conditions
[24]. These mechanisms of interneuronal spreading are currently of great research interest, and some evidence suggests the involvement of activity-dependent secretion by exosomes
[25] and/or chaperone-mediated pathways
[26].
All of these misfolded and aggregated proteins cause dysfunction and loss of synapses and eventually lead to the death of neurons
[12]. Various misfolded proteins are known to cause neurotoxicity, although the exact mechanisms are not yet clear. However, they can act both by toxic gain-of-function and loss-of-normal function
[12]. For instance, it has been shown that amyloid β (Aβ), tau and α-synuclein interfere with synaptic signaling
[27][28][29]. The mutant tau also disrupts microtubule function and neuronal transport mechanisms, while α-synuclein additionally disrupts mitochondrial protein import
[29].
In addition to synaptic dysfunction, one of the most prominent hallmarks of neurodegenerative disorders is cellular distress, characterized by the impairments of mitochondrial function, overproduction of reactive oxygen species, disrupted signaling cascade, and consequent neuroinflammation
[12]. These symptoms and the accumulation of misfolded proteins have a bidirectional relationship that is often mutually exacerbating
[12]. Aβ, α-synuclein and mutant huntingtin (mHtt) have been shown to induce acute oxidative stress in neurons and reduce the antioxidant capacity of astroglia
[30][31][32][33], while conversely, oxidative stress facilitates the aggregation of misfolded proteins and leads to proteostatic breakdown
[34][35]. Moreover, both Aβ oligomers and Aβ aggregates stimulate a low level of chronic neuroinflammation by activating microglia and astrocytes
[36]. These pro-inflammatory effects of Aβ, in turn, impair microglial and astroglial function as well as their ability to remove Aβ and other misfolded proteins
[31][36][37][38]. Finally, the overall process of neuroinflammation caused by misfolded proteins is likely exacerbated by age-related immune system senescence
[39][40].
3. Endocannabinoid System (ECS)
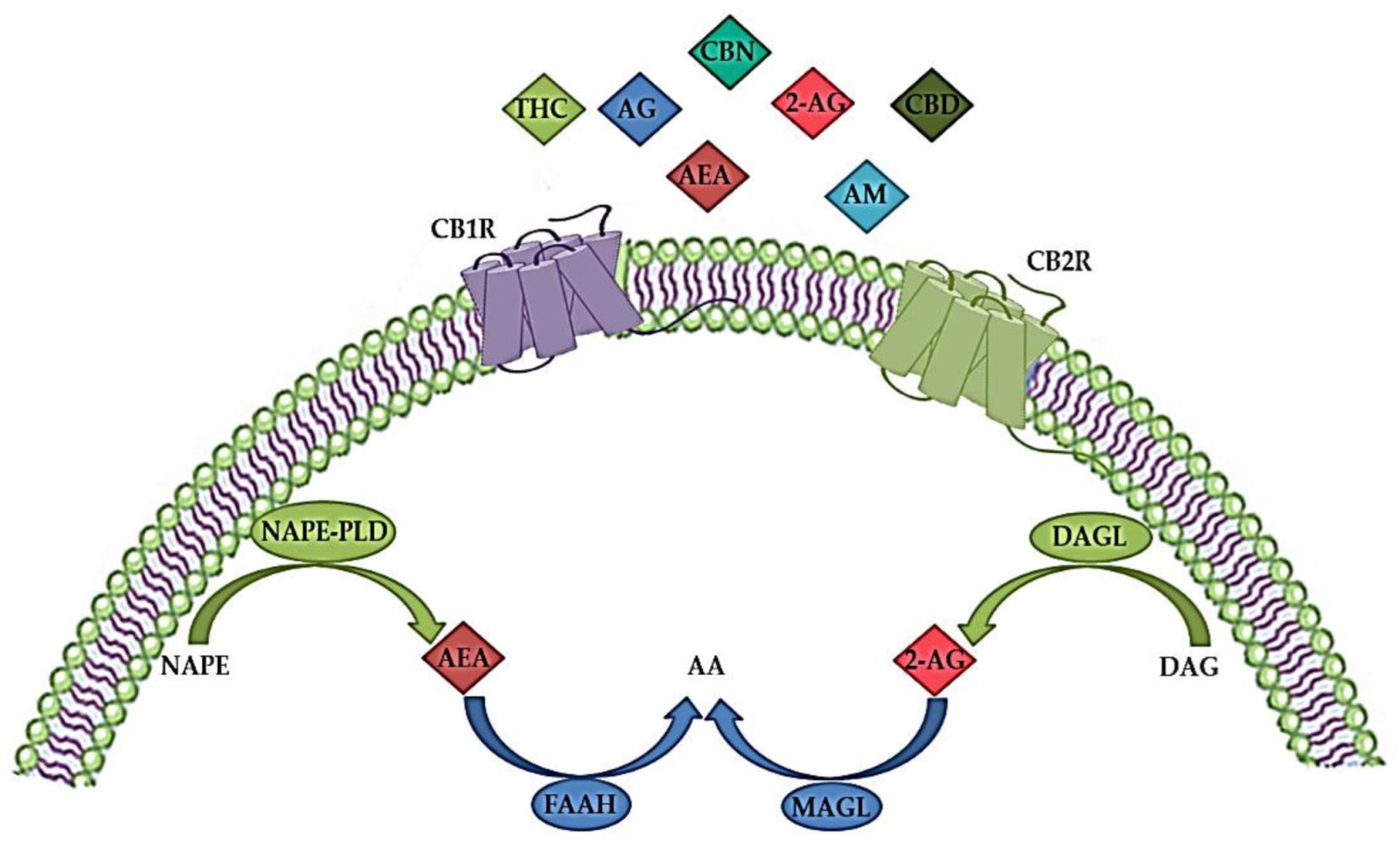
The ECS plays an important role in both the CNS and peripheral nervous system by modulating the neuronal network function and activity
[9]. It is a complex molecular system involved in various biological processes such as maintenance of homeostasis, neurogenesis, neuroprotection, cognition, pain, inflammation, learning and memory, as well as pre- and postnatal development
[41][42]. The ECS consists of endogenous cannabinoids (endocannabinoids), cannabinoid receptors (CBR), enzymes and other different proteins important for the transport and metabolism of endocannabinoids (
Figure 1)
[34][43]. Endocannabinoids are endogenous signaling lipid mediators that activate CBR and mimic the actions of Δ9-tetrahydrocannabinol (THC)
[43]. The biological effects of endocannabinoids are mediated by two members of the large family of G-protein-coupled receptors (GPCR); CB1R and CB2R (
Figure 1)
[29][44].
Figure 1. The endocannabinoid system (ECS) consists of endogenous cannabinoids (endocannabinoids) such as anandamide (arachidonoylethanolamide, AEA) and 2-arachidonoylglicerol (2-AG), anabolic enzymes (N-acylphosphatidylethanolamine-hydrolyzing phospholipase D (NAPE-PLD) and 1,2-diacylglycerol lipase (DAGL)), catabolic enzymes (fatty acid amide hydrolase (FAAH) and monoacylglycerol lipase (MAGL)) and cannabinoid receptors 1 (CBR1) and 2 (CB2R). In addition to endocannabinoids, various exogenous natural cannabinoids (phytocannabinoids), such as tetrahydrocannabinol (THC), cannabidiol (CBD) and cannabinol (CBN), as well as synthetic cannabinoids, such as different agonists (AG) or allosteric modulators (AM) act via CBR1 and/or CB2R. AA—arachidonic acid; DAG—diacylglycerol; NAPE—N-acylphosphatidylethanolamine. The image was created using Microsoft PowerPoint 2016.
These receptors are characterized by different signaling mechanisms, tissue distributions and differential expression in neurons and microglia
[45]. CB1R are mainly found in the CNS, in regions responsible for motor coordination (cerebellum, substantia nigra, striatum and basal ganglia), cognitive functions (cortex), as well as learning and memory (amygdala and hippocampus), but are also localized in the heart, uterus, testes, liver, gastrointestinal tract, immune cells and adipose tissue
[46][47]. In the CNS, CB1R are located in the presynaptic terminals of y-aminobutyric acid (GABA)-ergic, glutamatergic, cholinergic, noradrenergic and serotonergic neurons, and they regulate retrograde suppression of neurotransmission
[48]. Their distribution suggests an important role of these receptors in the regulation of cognition, memory and learning processes, movement, and emotions
[49], as well as in various neuropsychiatric disorders
[48][50]. CB2R are mainly found at the periphery, for instance, in the immune and hematopoietic system, but are activated in the CNS during inflammation, especially in microglia and astrocytes, as well as in oligodendrocytes, neural progenitor cells, and in the endothelium of the blood–brain barrier (BBB)
[51], suggesting their immunomodulatory role
[49]. Human CB2R has two isoforms; the CB2A isoform is expressed in the testes and brain, while the CB2B isoform is localized in the spleen and leukocytes
[52].
Both CB1R and CB2R are seven transmembrane domain receptors coupled to G-proteins (
Figure 1). They inhibit adenylyl cyclase, protein kinase A (PKA), and various voltage-gated calcium channels such as N-type, P/Q-type and L-type calcium currents and activate mitogen-activated protein kinases (MAPK) (including extracellular signal-regulated kinases (ERK), c-Jun N-terminal kinases (JNK), and p38 kinases) and inwardly rectifying potassium channels
[45][53]. Activation of CB1R blocks the release of various excitatory and inhibitory neurotransmitters and regulates the activity of specific ion channels
[54]. In addition, binding to CB1R stimulates signaling pathways such as phosphoinositide 3-kinase (PI3K)/Akt, MAPK and Nrf2 cascades involved in antioxidative defense and survival and activates N-methyl-D-aspartate (NMDA) receptors, Ca
2+ signaling cascades and influx, thereby regulating glutamatergic signaling
[55]. CB2R signaling also suppresses adenylyl cyclase, lowers cAMP levels, and decreases PKA activity
[55]. However, cAMP synthesis and activation of Akt and ERK signaling pathways are stimulated by CB2R signaling, probably by regulating different adenylyl cyclase isozymes
[54]. The protein kinase C (PKC) pathway, Janus kinase (JAK)/signal transducer and activator of transcription 1 (STAT1) pathway are some of the signaling pathways through which microglial activation is suppressed by CB2R stimulation
[56][57].
CBR activation is mediated by the two most common endogenous endocannabinoid ligands, anandamide (arachidonoylethanolamide, AEA) and 2-arachidonoylglicerol (2AG), both of which are derivates of n-6 polyunsaturated (PUFA) arachidonic acid (
Figure 1). AEA has high partial agonist affinity to CB1R but low efficacy at CB1R and even lower efficacy at CB2R, whereas 2-AG has low to moderate full agonist affinity to these two receptors but is fully effective
[34][58][59]. In addition, AEA is a full agonist at transient receptor potential cation channel subfamily V member 1 (TRPV1), also known as vanilloid receptor 1 (VR1), and at nuclear peroxisome proliferator-activated receptor (PPAR), while 2-AG binds to specific GABA receptor A subtypes in neuronal cells
[45][60].
High levels of AEA have been found in different brain regions, such as the hippocampus, thalamus, cerebral cortex, and cerebellum, while lower levels of AEA have been detected in the periphery, including human blood and cerebrospinal fluid
[61][62]. On the other hand, 2-AG is present in high concentrations in the brain stem, hippocampus, and striatum
[61]. Although AEA and 2-AG have different receptor affinities, synthesis, transport and inactivation pathways, both are produced in response to high intracellular Ca
2+ concentrations
[63][64]. As shown in
Figure 1, biosynthesis of AEA from N-arachidonoyl phosphatidylethanolamine by multiple pathways is initiated by a postsynaptic depolarization and an increase in Ca
2+ ions that activates N-acylphosphatidylethanolamine-hydrolyzing phospholipase D (NAPE-PLD) and diacylglycerol (DAG) lipase
[65]. 2-AG is synthesized from arachidonic acid-containing DAG by the action of DAG lipase (
Figure 1). DAG lipase alpha is important for the synaptic production of 2-AG in the adult brain, while DAG lipase beta is responsible for the microglial production of 2-AG
[66]. Studies have shown that disrupted synaptic localization of DAG lipase alpha is associated with CNS disorders
[67].
Due to their uncharged hydrophobic nature, endocannabinoids cannot diffuse easily like other neurotransmitters. There are three different models for the transport of AEA once it is released into the intracellular space
[68]: simple diffusion facilitated by the concentration gradient
[69], transport by protein carriers, and endocytosis
[70]. It is believed that 2-AG has a similar transport pathway, but it is not yet well described
[71]. Endocannabinoids taken up by cells can be degraded by two different pathways, hydrolysis and oxidation
[68]. Enzymes that are involved in the hydrolysis pathway include fatty acid amide hydrolase (FAAH) for AEA and monoacylglycerol lipase (MAGL) for 2-AG (
Figure 1)
[66][68][72]. The oxidation of both AEA and 2-AG involves cyclooxygenase (COX) and lipoxygenase (LOX)
[68].
ECS dysfunction and its alterations in the CNS are involved in the pathophysiology of neurodegenerative diseases such as Alzheimer’s disease
[73], Parkinson’s disease
[74], Huntington’s disease
[75], multiple sclerosis
[76] and amyotrophic lateral sclerosis
[48]. In addition, ECS dysregulation has been found in patients with schizophrenia
[77], anxiety disorders
[78] and major depressive disorder (MDD)
[79]. Neuroimmune and neurooxidative pathways are involved in neurocognitive impairments and various behavioral symptoms, as observed in several neuropsychiatric disorders
[79][80][81]. Therefore, due to their neuroprotective and neuroinflammatory roles in the CNS, pharmacological modulation of different components of the ECS may have therapeutic potential in various CNS disorders
[82].
4. Modulation of Cannabinoid Receptor 2 (CB2R)
Marijuana or cannabis (
Cannabis sativa) contains about 500 compounds, of which at least 100 are classified as phytocannabinoids with different chemical structures and pharmacological properties, the most abundant natural cannabinoids being THC, cannabidiol (CBD) and cannabinol (CBN)
[83]. CBR1 and CRBR2 were first studied as targets of THC in the human brain
[84]. THC interacts with both CBR, as an agonist at CB1R and as a weak antagonist at CB2R
[85], but also possibly by inhibiting COX enzymes and as an inducer of COX-2 with prolonged exposure
[86]. In addition, another natural cannabinoid CBD, which has a low affinity for CBR, may be a CB2R inverse agonist with anti-inflammatory effects
[87]. Subsequently, the identification of CBR in the brain suggested the presence of endogenous ligands, and the most studied and characterized endocannabinoids are AEA and 2-AG, which have an affinity for both CBR
[84][88]. It was found that 2-AG acts as a full agonist, while AEA acts as a weak partial agonist for both CB1R and CB2R
[85].
CP 55,940 was the first synthetic cannabinoid analog to be synthesized, followed by several others. Although many synthetic cannabinoids are known to have an affinity for both CB1R and CB2R, they, like natural cannabinoids, can also interact with non-CBR, such as vanilloid or serotonergic receptors
[89]. The most common group of synthetic cannabinoids is JWH, where JWH-018 has potent pharmacological activity and can be easily synthesized and used to synthesize other synthetic cannabinoids with different properties and affinities for CBR
[90]. Besides JWH, other common groups of synthetic cannabinoids are HU and CP groups. While HU are classic cannabinoids, CP are cannabimimetics originally developed by Pfizer in the 1970s
[90]. Some of the most extensively studied selective CB1 or mixed CB1/CB2 agonists are WIN 55,212-2, HU 210, ACEA, and JWH-018
[91]. In addition, several synthetic selective CB2R agonists, such as GSK554418A, GW833972A, GW842166X, HU-308, GW405833, JWH-015, JWH-133, A-836339, AM1241, AM630, NESS400, etc., have been reported in the literature and some (Cannabinir, GW842166, Tedalinab, GRC10693, S-7774698, LY2828360, KHK6188, Lenabasum) are under investigation at various stages of clinical development
[85][89].
In humans, CB2R are encoded by the cannabinoid receptor 2 (
CNR2) gene, which is located on chromosome 1p36 and consists of 360 amino acids
[92]. The CB2R share 44% total amino acid homology and 68% homology in the transmembrane domains with the CB1R
[92]. The CB2R were cloned in 1993, and these receptors were previously thought to be absent from the brain, as they were only detectable in the periphery
[51][93][94].
In contrast to CB1R, which are mainly found in the CNS, particularly in presynaptic neurons at central and peripheral nerve terminals, where they inhibit neurotransmitter release
[95], CB2R predominate in cells and tissues involved in the immune response, such as the spleen, thymus and blood-derived monocytes
[51][96], and modulate interleukin release and cell migration.
Until recently, the significant increase of CB2R in the CNS was thought to occur specifically in activated microglial cells under inflammatory conditions but was not measurable under physiological conditions or in other brain cell types
[97]. However, using methods such as immunostaining, in situ hybridization, and gene expression analysis, CB2R has been shown to be present throughout different brain regions
[98][99][100][101], including the striatum, amygdala, hippocampus, cortex and ventral tegmental area
[102], in neural progenitor cells, neurons, as well as glial and endothelial cells
[103][104][105][106]. In neurons, CB2Rs appear to be mainly distributed in postsynaptic somatodendritic regions, and their activation inhibits neuronal excitability through membrane hyperpolarization
[97][98][101]. Novel detection techniques allowed more precise detection of low CB2R mRNA levels, specifically in astrocytes, dopaminergic, glutamatergic and GABAergic neurons, but not in resting microglia
[11][107][108][109].
Although there are numerous studies on the regulation of CB1R, knowledge of the physiological and pathological role of CB2R is limited. Activation of CB2R leads to the inhibition of neuroinflammatory signaling pathways, as well as a return from the pro-inflammatory state of microglia to normal anti-inflammatory function
[85]. Thus, in in vitro experiments, AEA has been shown to act via the MAPK signaling pathway within the CNS immune system to reduce the magnitude of the inflammatory response, as well as to limit neurodegenerative immune responses
[110]. Moreover, AEA was found to reduce lipopolysaccharide-induced neuroinflammation in primary rat microglial cultures
[111]. Even though AEA can activate CB1R, CB2R and other receptors of the ECS, the anti-inflammatory actions appear to be mediated by CB2R
[112]. Therefore, AEA may have a potential therapeutic effect on microglial-derived neuroinflammation and regulate many aspects of the inflammatory response in the brain. However, since CB2R ligands exert neuroprotection without psychotropic effects (strong mood alterations, anxiety, acute psychosis, cognitive and motor impairments), usually seen with CB1R agonists
[95], new and selective CB2R ligands may be promising and safe drugs for the treatment of various neuroinflammatory disorders
[111]. Nevertheless, CB2R agonists also have disadvantages, such as immune suppression during chronic use, or pro-inflammatory actions
[111].
Only a few synthetic CB2R agonists have reached clinical trials, despite increasing reports of selective CB2R ligands and high expectations with these ECS targets
[113]. Some of them, such as GW842166X, CP55940, S-777469 and JTE-907, have already completed Phase II trials in various pain disorders; however, none of them have been assessed for neurodegenerative or neuroinflammatory disorders in humans
[111]. Recently, new CB2R ligands have been characterized for their potential neuroprotective effects and the most prominent among them, the inverse agonist of CB2R SMM-189 seems to achieve neuroprotection by modulating microglial activation in a mouse model of mild traumatic brain injury
[114]. Specifically, SMM-189 reduces some pro-inflammatory markers, indicating decreased infiltration of peripheral macrophages and other immune cells involved in neurodegeneration
[115].
Recently, new strategies targeting CB2R for neurodegenerative and neuroinflammatory disorders have emerged when 4′-O-methylhokiol, the main bioactive component of Magnolia grandiflora L. that acts as both CB2R modulator and COX-2 substrate-specific inhibitor, has shown beneficial effects in animal models of neurodegeneration
[116]. Moreover, targeting CB2R homo- and heterodimers needs to be further investigated
[111]. While homobivalent and heterobivalent CB1R ligands have been previously designed and described in the literature
[117], the first structurally bivalent CB2R compounds were designed and synthesized in 2014; however, with lower activity and selectivity compared to their monomeric counterparts
[118]. Whereas monomeric compound is a selective CB2R agonists, bivalent compounds are weak antagonists/inverse agonists at CB1R and CB2R
[118].
Another therapeutic possibility is the use of ligand-biased signaling
[119]. For example, 2-AG is a very potent activator of the ERK1/2-MAPK signaling pathway at low concentrations, although higher concentrations are required to inhibit the adenylyl cyclase and calcium pathways
[120]. In the future, CB2R allosteric modulators may offer new therapeutic approaches due to their potential to fine-tune receptor responses while minimizing the side effects
[111]. Currently, the allosteric modulation specific to the CB2R signaling is still evaluated
[121], while CB2R positive and negative allosteric modulators remain to be discovered.
5. Role of CB2R in Various CNS Disorders
Neurodegenerative disorders, such as Alzheimer’s disease, Parkinson’s disease, Huntington’s disease, multiple sclerosis and amyotrophic lateral sclerosis, are characterized by a progressive loss of specific neurons in various brain regions, which leads to different symptomatic and clinical outcomes
[122][123]. Since there is no cure for these diseases, therapeutic approaches mostly consist of partial symptomatic relief, which do not halt the progression of the disease. The main hallmarks of neurodegenerative diseases are neuroinflammation, oxidative stress, abnormal protein accumulation and excitotoxicity
[124]. Studies have shown that pharmacological modulation of endocannabinoid signaling can modulate these neurodegeneration traits and cause alleviation of symptoms and disease progression
[125]. Targeting different components of ECS, therefore, brings new aspects to understanding mechanisms underlying different CNS disorders to provide novel, more effective therapies.
The role of CB1R in behavior, emotions, learning and memory, addiction and various other CNS disorders has been widely studied
[49][126]. Previous studies have shown that dysregulation of CB1R in different CNS regions is involved in the pathophysiology of schizophrenia, MDD and anxiety disorders
[127][128][129]. Therefore, normalizing the CB1R activity may have beneficial effects in treating these disorders. However, obtained findings demonstrated that CB1R pharmacological targeting induces serious side effects, such as depression, psychosis, panic attacks, anxiety and even suicidal ideation
[130][131]. Hence, there is an emerging need to study new therapeutic targets with minimal adverse effects
[130][131]. Recent studies suggested that targeting CB2R in CNS is effective and safe and may open a new possibility for the modulation of ECS.
Compared to CB1R, CB2R have lower expression levels in the brain under normal physiological conditions, but their enhanced levels were observed in neurodegenerative and neuropsychiatric disorders
[101][105][132]. Recent findings suggested that B2R modulates the behavioral effects in the CNS
[9], including mood and emotional behavior. Evidence suggests CB2R plays a role in food intake, body weight control and eating disorders
[133][134][135], depression and anxiety
[101][105][134], drug addiction
[136], psychosis and schizophrenia-like behavior
[137][138][139] and synaptic plasticity underlying cognitive functions
[135][138].
Elevated CB2R expression levels have been reported in several pathological conditions, such as neurological pain
[136][140], stroke
[137], traumatic brain injury
[140][141], addiction
[142][143] and neurodegenerative diseases, including multiple sclerosis
[144][145]. CB2R anti-inflammatory action has been found in animal studies and in experiments using cell cultures
[141][146]. The activation of CB2R decreases neuroinflammation, partly by mediating the transition of microglial phenotype from a predominantly neurotoxic ‘’M1” to a neuroprotective ‘’M2”
[147], suggesting an important role of CB2R in restoring homeostasis
[85]. Therefore, due to CBR2 inducible nature during inflammation, ligands that activate or inhibit their activity could be used for potential therapeutic purposes in various CNS disorders whose pathogenesis involves neuroinflammatory processes
[97].
6. Role of CB2R in Neuroinflammation and Neurodegeneration
A strong relationship between neuroinflammation and neurodegeneration has been reported in the early stages of neurodegenerative disorders, such as Alzheimer’s disease, frontotemporal dementia, Parkinson’s disease, amyotrophic lateral sclerosis and Huntington’s disease. This link is also strong in primarily inflammatory diseases such as multiple sclerosis and human immunodeficiency virus (HIV)-associated dementia associated with intense and chronic inflammation of myelin sheets and HIV infection of microglia, respectively, consequently leading to neuronal damage
[123]. Moreover, the neuroinflammatory condition is characteristic of other psychiatric disorders and neurological diseases such as epilepsy, and traumatic brain injury, where it mediates secondary neurodegeneration
[148][149].
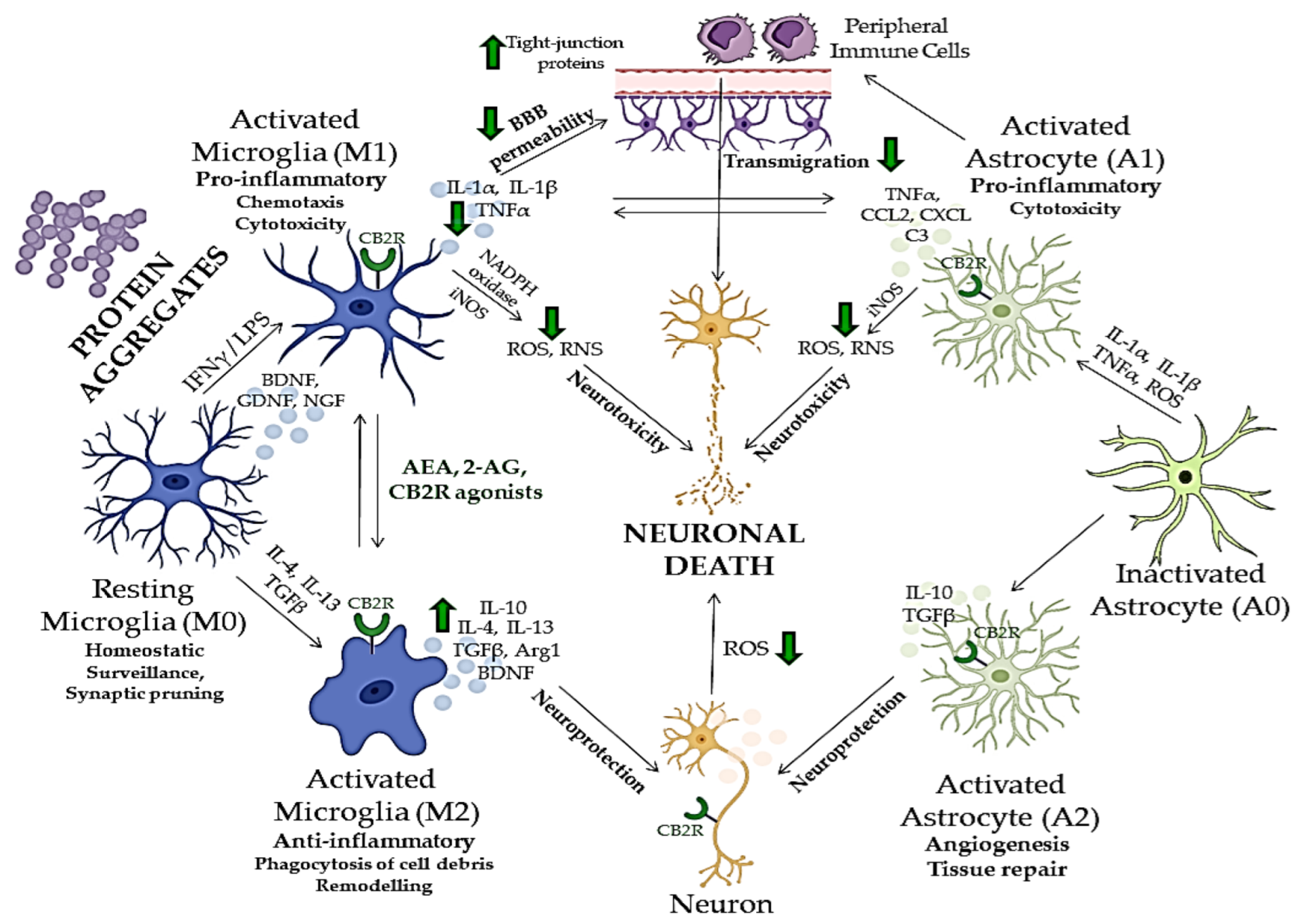
Many aspects of neuroinflammation and neurodegeneration cross-talk remain unclear; however, recent studies showed that glial cells, especially microglia, which act as the brain’s immune cells, could be crucial mediators of neurodegeneration, together with peripheral monocytes which cross BBB under CNS pathological conditions (
Figure 2)
[150][151][152]. ECS has been, therefore, extensively investigated in relation to neurodegenerative and neuroinflammatory mechanisms of CNS disorders, and potentially novel treatment strategies
[153]. Although there are major differences in the etiology, physiology and clinical picture of various neurodegenerative proteinopathies, aggregation and accumulation of defected and misfolded proteins such as Aβ, hyperphosphorylated tau
[154], α-synuclein
[155], mutated superoxidase dismutase 1 (mSOD1)
[156] and huntingtin
[157], are shared aspects of these disorders and all represent activation stimulus to circulating microglia
[158].
Figure 2. The cross-talk between neuroinflammation and neurodegeneration and neuroprotective effects of compounds acting via CB2R. Green arrows indicate the effects of endocannabinoids or CB2R agonists on different mediators of neuroinflammation and neurodegeneration. BBB—blood–brain barrier; CB2R—cannabinoid receptor 2; ROS—reactive oxygen species; RNS—reactive nitrogen species; iNOS—inducible nitric oxide synthase; IFNγ—interferon gamma, LPS—lipopolysaccharide; IL—interleukin; TGFβ—tumor growth factor beta; TNFα—tumor necrosis factor alpha; BDNF—brain-derived neurotrophic factor; NGF—neuron growth factor; GNDF—glial cell-derived neurotrophic factor; NADPH—nicotinamide adenine dinucleotide phosphate; Arg1—arginase 1; CCL2—C-C motif chemokine ligand 2; CXCL—C-X-C motif chemokine ligand; C3—complement C3. The image was created using Microsoft PowerPoint 2016.
Glial cells play a central role in inducing and maintaining neuronal synaptic plasticity and represent the first line of defense against neuroinflammation
[141][159]. Microglia typically occur in three states, distinguished by their receptor expression profile, morphology, and biological functions
[11]. Inactivated microglia (M0) are characteristic of homeostatic, non-pathological conditions. Their role is scanning the environment for potential infectious components
[160] and regulating the growth and protrusion of dendritic spines
[161]. In response to various CNS insults
[162], microglia transit from an anti-inflammatory to a reactive pro-inflammatory phenotype (M1), which exhibits cytotoxic and phagocytic activity to eliminate damaged neurons and cellular debris
[163]. The activation of resting microglia, as an answer to a threat, leads to microglial polarization, which results in exacerbated neuroinflammation, excitotoxicity and oxidative stress
[150]. M1 microglia is characterized by the production and secretion of ROS and reactive nitrogen species (RNS), inflammatory cytokines such as tumor necrosis factor-α (TNF-α), interleukin 1β (IL-1β), IL-6 or IL-12 and recruitment of other immune cells
[164][165]. After the threat is gone, M1 microglia switches to an alternative activation state (M2) in which it produces anti-inflammatory and neuroprotective factors, such as IL-10, tumor growth factor-β (TGF-β) and brain-derived neurotrophic factor (BDNF) to dampen further inflammation and induce healing process
[165]. However, chronic activation of inflammatory signaling pathways and continuous release of inflammatory cytokines and chemokines by reactive microglia can subsequently result in damage to neurons
[166][167], which underlies the pathogenesis of various neurodegenerative disorders (
Figure 2)
[85].
CBR expression changes over time in the brain and at the periphery, depending on the different stages of neurodegeneration
[168]. During neurodegeneration, CB1R-expressing neurons show a progressive loss. For instance, in the early stages of Alzheimer’s disease, the activity of CB1R is increased in the hippocampus, whereas in its advanced stages, decreased CB1R activity has been observed
[168]. On the other hand, in a healthy brain, CB2R expression is modest, but it rises in activated astrocytes and microglia
[168]. In particular, activated microglia show both increased CB2R expression and higher endocannabinoid synthesis
[169]. Upregulated endocannabinoid signaling alleviates microglial over-activation, inhibits pro-inflammatory cytokine release, reduces microglial overactivity, and decreases phagocytic capability
[92][170]. One of the main mechanisms by which CB2R might counteract neuroinflammation and attenuate neurodegeneration is changing the microglial polarization, namely shifting it to protective and anti-inflammatory (M0 and M2) states (
Figure 2)
[169]. For instance, CB2R activation by AEA through activation of ERK1/2 and JNK boosted the expression of anti-inflammatory cytokines (IL-10), characteristic for M2 microglia, and decreased the expression of M1 characteristic markers, while CB2R inhibition inhibited this effect
[171]. Additionally, the polarization to the M2 state is disrupted by
CB2R deletion in microglial cells from
CB2R knockout mice
[169].
Besides microglia, neuronal homeostasis could be maintained by CB2R activation in neurons, leading to a reduction of oxidative damage by influencing the expression of neuronal nitric oxide synthase (NOS), excitotoxicity and apoptosis
[172][173]. In astrocytes, which express both CB1R and CB2R, ECS activation leads to the simultaneous production of anti-inflammatory factors and inhibition of pro-inflammatory cytokines, and to lower inducible NOS (iNOS) expression and decreased release of neurotoxic factors
[92][174][175]. Additionally, it has been demonstrated that CB2R activation in brain microvascular endothelial cells reduces the tight junction protein expression and BBB permeability after traumatic brain injury, which prevents peripheral immune cells from migrating further into the CNS
[176].
Nevertheless, it is reasonable to consider microglia as one of the central factors underlying neurodegenerative pathology, both in a protective and toxic manner
[177]. In neurodegenerative proteinopathies, the primary role of activated microglia is the clearance of misfolded proteins and damaged cells, which is followed by a healing phase and neuroprotection. However, the progressive nature of these diseases and the ongoing production of misfolded proteins, continuously activate the cytotoxic state of microglia, which leads to the overly activated inflammatory response and diminished healing possibilities, thereby worsening the clinical picture
[177]. ECS and CB2R have been shown to regulate the neuroinflammation in neurodegenerative disorders, mostly in microglia, but also through neuronal and astroglial cells and their cross-talk (
Figure 2)
[58][168]. Additionally, numerous research studies have shown that both endogenous and exogenous cannabinoids lower the microglial over-activation and effectively ameliorate the neurotoxic effects and neurodegeneration in various neuropsychiatric disorders (
Figure 2)
[9].
 +1 credit
+1 credit