Some of the most prevalent neurodegenerative disorders, including Alzheimer’s and Parkinson’s disease, are proteinopathies characterized by the accumulation of specific protein aggregates in the brain. Such misfolded protein aggregates can trigger modulation of the innate and adaptive immune systems and subsequently lead to chronic neuroinflammation that drives the onset and progression of neurodegenerative diseases. Since there is still no effective disease-modifying treatment, new therapeutic targets for neurodegenerative proteinopathies have been sought. The endocannabinoid system, and in particular the cannabinoid CB2 receptors, have been extensively studied, due to their important role in neuroinflammation, especially in microglial cells.
- neurodegenerative diseases
- proteinopathies
- endocannabinoid system
- cannabinoid CB2 receptors
1. Introduction
2. Neurodegenerative Proteinopathies
Neurodegenerative proteinopathies is an umbrella term for neurodegenerative disorders characterized by the formation of misfolded protein aggregates that cause cellular toxicity and contribute to cellular proteostatic collapse [13][12]. According to the pathophysiological hypothesis of neurodegenerative disorders, some proteins change their conformations, consequently gaining neurotoxic activity or losing their normal function by forming small oligomeric or large fibrillar aggregates, leading to neurodegeneration [3]. Neurodegenerative proteinopathies include some of the most common neurodegenerative disorders, such as Alzheimer’s and Parkinson’s disease, as well as Huntington’s disease, multiple sclerosis, amyotrophic lateral sclerosis, etc. The recent findings showing that bassoon proteinopathy drives neurodegeneration in mice and patients with multiple sclerosis are reminiscent of disease pathways in neurodegenerative proteinopathies [12][13]. The neuronal proteome consists of 10,000 to 20,000 different proteins that, in order to fulfill their biological function, must fold in accordance with the instructions encoded in the amino acid sequence [14]. Therefore, to maintain cellular integrity and health, the process of protein folding and its degradation must be well-regulated [15]. However, their biologically active conformation (the native state) is often marginally stable under normal physiological conditions, and even a small polypeptide of ~100 amino acids can adopt many conformations (~1030) under different conditions [14]. It is, therefore, hardly surprising that the process of protein folding is error-prone, leading to misfolded states and off-pathway aggregates [14]. Due to this susceptibility, cells face a continuous stream of misfolded and aggregated proteins (Table 1) and require supportive molecular chaperones called heat shock proteins (Hsp) to refold, degrade, and eliminate them to maintain proteome homeostasis [13][12].| Neurodegenerative Proteinopathy | Misfolded and Aggregated Protein(S) | ||
|---|---|---|---|
| Alzheimer’s disease (AD) | Amyloid beta (Aβ) peptide, tau | ||
| Parkinson’s disease (PD) | α-synuclein | ||
| Huntington´s disease (HD) | Mutant huntingtin (mHtt) | ||
| Multiple sclerosis (MS) | Bassoon presynaptic cytomatrix protein | ||
| Amyotrophic lateral sclerosis (ALS) | Mutant superoxide dismutase 1 (mSOD1), TAR DNA-binding protein 43 (TDP-43) and fused-in-sarcoma (FUS) protein | ||
| Dementia with Lewy bodies (DLB) | α-synuclein | ||
| Frontotemporal lobar degeneration (FTLD) | FTLD-tau, FTLD-TDP, FTLD-FUS | ||
| Creutzfeldt–Jakob disease (CJD) | Protease-resistant cellular prion protein (PrP | Sc | ) |
3. Endocannabinoid System (ECS)
The ECS plays an important role in both the CNS and peripheral nervous system by modulating the neuronal network function and activity [9]. It is a complex molecular system involved in various biological processes such as maintenance of homeostasis, neurogenesis, neuroprotection, cognition, pain, inflammation, learning and memory, as well as pre- and postnatal development [41,42][41][42]. The ECS consists of endogenous cannabinoids (endocannabinoids), cannabinoid receptors (CBR), enzymes and other different proteins important for the transport and metabolism of endocannabinoids (Figure 1) [34,43][34][43]. Endocannabinoids are endogenous signaling lipid mediators that activate CBR and mimic the actions of Δ9-tetrahydrocannabinol (THC) [43]. The biological effects of endocannabinoids are mediated by two members of the large family of G-protein-coupled receptors (GPCR); CB1R and CB2R (Figure 1) [29,44][29][44].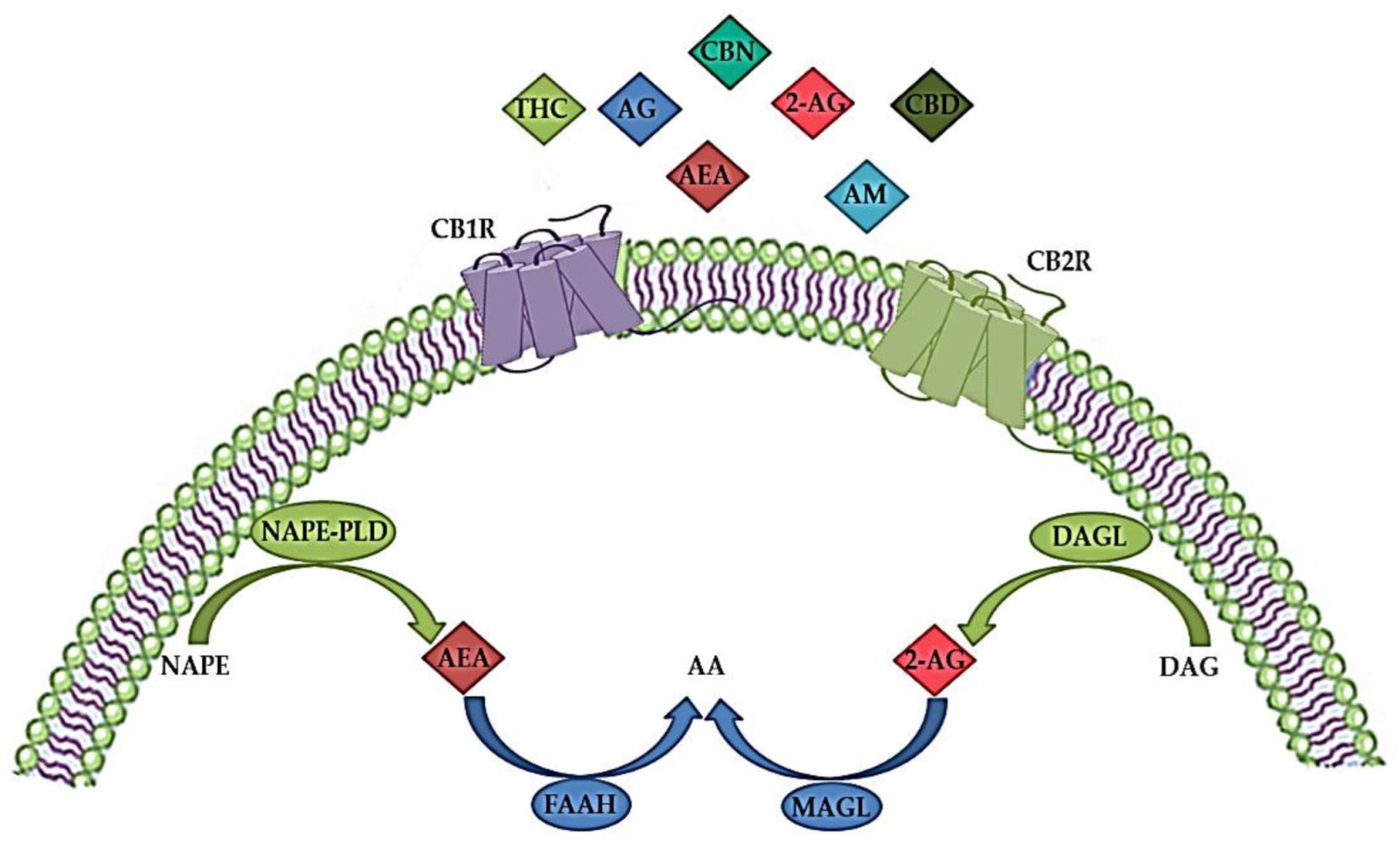
4. Modulation of Cannabinoid Receptor 2 (CB2R)
Marijuana or cannabis (Cannabis sativa) contains about 500 compounds, of which at least 100 are classified as phytocannabinoids with different chemical structures and pharmacological properties, the most abundant natural cannabinoids being THC, cannabidiol (CBD) and cannabinol (CBN) [83]. CBR1 and CRBR2 were first studied as targets of THC in the human brain [84]. THC interacts with both CBR, as an agonist at CB1R and as a weak antagonist at CB2R [85], but also possibly by inhibiting COX enzymes and as an inducer of COX-2 with prolonged exposure [86]. In addition, another natural cannabinoid CBD, which has a low affinity for CBR, may be a CB2R inverse agonist with anti-inflammatory effects [87]. Subsequently, the identification of CBR in the brain suggested the presence of endogenous ligands, and the most studied and characterized endocannabinoids are AEA and 2-AG, which have an affinity for both CBR [84,88][84][88]. It was found that 2-AG acts as a full agonist, while AEA acts as a weak partial agonist for both CB1R and CB2R [85]. CP 55,940 was the first synthetic cannabinoid analog to be synthesized, followed by several others. Although many synthetic cannabinoids are known to have an affinity for both CB1R and CB2R, they, like natural cannabinoids, can also interact with non-CBR, such as vanilloid or serotonergic receptors [89]. The most common group of synthetic cannabinoids is JWH, where JWH-018 has potent pharmacological activity and can be easily synthesized and used to synthesize other synthetic cannabinoids with different properties and affinities for CBR [90]. Besides JWH, other common groups of synthetic cannabinoids are HU and CP groups. While HU are classic cannabinoids, CP are cannabimimetics originally developed by Pfizer in the 1970s [90]. Some of the most extensively studied selective CB1 or mixed CB1/CB2 agonists are WIN 55,212-2, HU 210, ACEA, and JWH-018 [91]. In addition, several synthetic selective CB2R agonists, such as GSK554418A, GW833972A, GW842166X, HU-308, GW405833, JWH-015, JWH-133, A-836339, AM1241, AM630, NESS400, etc., have been reported in the literature and some (Cannabinir, GW842166, Tedalinab, GRC10693, S-7774698, LY2828360, KHK6188, Lenabasum) are under investigation at various stages of clinical development [85,89][85][89]. In humans, CB2R are encoded by the cannabinoid receptor 2 (CNR2) gene, which is located on chromosome 1p36 and consists of 360 amino acids [92]. The CB2R share 44% total amino acid homology and 68% homology in the transmembrane domains with the CB1R [92]. The CB2R were cloned in 1993, and these receptors were previously thought to be absent from the brain, as they were only detectable in the periphery [51,93,94][51][93][94]. In contrast to CB1R, which are mainly found in the CNS, particularly in presynaptic neurons at central and peripheral nerve terminals, where they inhibit neurotransmitter release [95], CB2R predominate in cells and tissues involved in the immune response, such as the spleen, thymus and blood-derived monocytes [51[51][96],96], and modulate interleukin release and cell migration. Until recently, the significant increase of CB2R in the CNS was thought to occur specifically in activated microglial cells under inflammatory conditions but was not measurable under physiological conditions or in other brain cell types [97]. However, using methods such as immunostaining, in situ hybridization, and gene expression analysis, CB2R has been shown to be present throughout different brain regions [98,99[98][99][100][101],100,101], including the striatum, amygdala, hippocampus, cortex and ventral tegmental area [102], in neural progenitor cells, neurons, as well as glial and endothelial cells [103,104,105,106][103][104][105][106]. In neurons, CB2Rs appear to be mainly distributed in postsynaptic somatodendritic regions, and their activation inhibits neuronal excitability through membrane hyperpolarization [97,98,101][97][98][101]. Novel detection techniques allowed more precise detection of low CB2R mRNA levels, specifically in astrocytes, dopaminergic, glutamatergic and GABAergic neurons, but not in resting microglia [11,107,108,109][11][107][108][109]. Although there are numerous studies on the regulation of CB1R, knowledge of the physiological and pathological role of CB2R is limited. Activation of CB2R leads to the inhibition of neuroinflammatory signaling pathways, as well as a return from the pro-inflammatory state of microglia to normal anti-inflammatory function [85]. Thus, in in vitro experiments, AEA has been shown to act via the MAPK signaling pathway within the CNS immune system to reduce the magnitude of the inflammatory response, as well as to limit neurodegenerative immune responses [110]. Moreover, AEA was found to reduce lipopolysaccharide-induced neuroinflammation in primary rat microglial cultures [111]. Even though AEA can activate CB1R, CB2R and other receptors of the ECS, the anti-inflammatory actions appear to be mediated by CB2R [112]. Therefore, AEA may have a potential therapeutic effect on microglial-derived neuroinflammation and regulate many aspects of the inflammatory response in the brain. However, since CB2R ligands exert neuroprotection without psychotropic effects (strong mood alterations, anxiety, acute psychosis, cognitive and motor impairments), usually seen with CB1R agonists [95], new and selective CB2R ligands may be promising and safe drugs for the treatment of various neuroinflammatory disorders [111]. Nevertheless, CB2R agonists also have disadvantages, such as immune suppression during chronic use, or pro-inflammatory actions [111]. Only a few synthetic CB2R agonists have reached clinical trials, despite increasing reports of selective CB2R ligands and high expectations with these ECS targets [113]. Some of them, such as GW842166X, CP55940, S-777469 and JTE-907, have already completed Phase II trials in various pain disorders; however, none of them have been assessed for neurodegenerative or neuroinflammatory disorders in humans [111]. Recently, new CB2R ligands have been characterized for their potential neuroprotective effects and the most prominent among them, the inverse agonist of CB2R SMM-189 seems to achieve neuroprotection by modulating microglial activation in a mouse model of mild traumatic brain injury [114]. Specifically, SMM-189 reduces some pro-inflammatory markers, indicating decreased infiltration of peripheral macrophages and other immune cells involved in neurodegeneration [115]. Recently, new strategies targeting CB2R for neurodegenerative and neuroinflammatory disorders have emerged when 4′-O-methylhokiol, the main bioactive component of Magnolia grandiflora L. that acts as both CB2R modulator and COX-2 substrate-specific inhibitor, has shown beneficial effects in animal models of neurodegeneration [116]. Moreover, targeting CB2R homo- and heterodimers needs to be further investigated [111]. While homobivalent and heterobivalent CB1R ligands have been previously designed and described in the literature [117], the first structurally bivalent CB2R compounds were designed and synthesized in 2014; however, with lower activity and selectivity compared to their monomeric counterparts [118]. Whereas monomeric compound is a selective CB2R agonists, bivalent compounds are weak antagonists/inverse agonists at CB1R and CB2R [118]. Another therapeutic possibility is the use of ligand-biased signaling [119]. For example, 2-AG is a very potent activator of the ERK1/2-MAPK signaling pathway at low concentrations, although higher concentrations are required to inhibit the adenylyl cyclase and calcium pathways [120]. In the future, CB2R allosteric modulators may offer new therapeutic approaches due to their potential to fine-tune receptor responses while minimizing the side effects [111]. Currently, the allosteric modulation specific to the CB2R signaling is still evaluated [121], while CB2R positive and negative allosteric modulators remain to be discovered.5. Role of CB2R in Various CNS Disorders
Neurodegenerative disorders, such as Alzheimer’s disease, Parkinson’s disease, Huntington’s disease, multiple sclerosis and amyotrophic lateral sclerosis, are characterized by a progressive loss of specific neurons in various brain regions, which leads to different symptomatic and clinical outcomes [122,123][122][123]. Since there is no cure for these diseases, therapeutic approaches mostly consist of partial symptomatic relief, which do not halt the progression of the disease. The main hallmarks of neurodegenerative diseases are neuroinflammation, oxidative stress, abnormal protein accumulation and excitotoxicity [124]. Studies have shown that pharmacological modulation of endocannabinoid signaling can modulate these neurodegeneration traits and cause alleviation of symptoms and disease progression [125]. Targeting different components of ECS, therefore, brings new aspects to understanding mechanisms underlying different CNS disorders to provide novel, more effective therapies. The role of CB1R in behavior, emotions, learning and memory, addiction and various other CNS disorders has been widely studied [49,126][49][126]. Previous studies have shown that dysregulation of CB1R in different CNS regions is involved in the pathophysiology of schizophrenia, MDD and anxiety disorders [127,128,129][127][128][129]. Therefore, normalizing the CB1R activity may have beneficial effects in treating these disorders. However, obtained findings demonstrated that CB1R pharmacological targeting induces serious side effects, such as depression, psychosis, panic attacks, anxiety and even suicidal ideation [130,131][130][131]. Hence, there is an emerging need to study new therapeutic targets with minimal adverse effects [130,131][130][131]. Recent studies suggested that targeting CB2R in CNS is effective and safe and may open a new possibility for the modulation of ECS. Compared to CB1R, CB2R have lower expression levels in the brain under normal physiological conditions, but their enhanced levels were observed in neurodegenerative and neuropsychiatric disorders [101,105,132][101][105][132]. Recent findings suggested that B2R modulates the behavioral effects in the CNS [9], including mood and emotional behavior. Evidence suggests CB2R plays a role in food intake, body weight control and eating disorders [133,134[133][134][135],135], depression and anxiety [101[101][105][134],105,134], drug addiction [136], psychosis and schizophrenia-like behavior [137,138,139][137][138][139] and synaptic plasticity underlying cognitive functions [135,138][135][138]. Elevated CB2R expression levels have been reported in several pathological conditions, such as neurological pain [136[136][140],140], stroke [137], traumatic brain injury [140[140][141],141], addiction [142,143][142][143] and neurodegenerative diseases, including multiple sclerosis [144,145][144][145]. CB2R anti-inflammatory action has been found in animal studies and in experiments using cell cultures [141,146][141][146]. The activation of CB2R decreases neuroinflammation, partly by mediating the transition of microglial phenotype from a predominantly neurotoxic ‘’M1” to a neuroprotective ‘’M2” [147], suggesting an important role of CB2R in restoring homeostasis [85]. Therefore, due to CBR2 inducible nature during inflammation, ligands that activate or inhibit their activity could be used for potential therapeutic purposes in various CNS disorders whose pathogenesis involves neuroinflammatory processes [97].6. Role of CB2R in Neuroinflammation and Neurodegeneration
A strong relationship between neuroinflammation and neurodegeneration has been reported in the early stages of neurodegenerative disorders, such as Alzheimer’s disease, frontotemporal dementia, Parkinson’s disease, amyotrophic lateral sclerosis and Huntington’s disease. This link is also strong in primarily inflammatory diseases such as multiple sclerosis and human immunodeficiency virus (HIV)-associated dementia associated with intense and chronic inflammation of myelin sheets and HIV infection of microglia, respectively, consequently leading to neuronal damage [123]. Moreover, the neuroinflammatory condition is characteristic of other psychiatric disorders and neurological diseases such as epilepsy, and traumatic brain injury, where it mediates secondary neurodegeneration [148,149][148][149]. Many aspects of neuroinflammation and neurodegeneration cross-talk remain unclear; however, recent studies showed that glial cells, especially microglia, which act as the brain’s immune cells, could be crucial mediators of neurodegeneration, together with peripheral monocytes which cross BBB under CNS pathological conditions (Figure 2) [150,151,152][150][151][152]. ECS has been, therefore, extensively investigated in relation to neurodegenerative and neuroinflammatory mechanisms of CNS disorders, and potentially novel treatment strategies [153]. Although there are major differences in the etiology, physiology and clinical picture of various neurodegenerative proteinopathies, aggregation and accumulation of defected and misfolded proteins such as Aβ, hyperphosphorylated tau [154], α-synuclein [155], mutated superoxidase dismutase 1 (mSOD1) [156] and huntingtin [157], are shared aspects of these disorders and all represent activation stimulus to circulating microglia [158].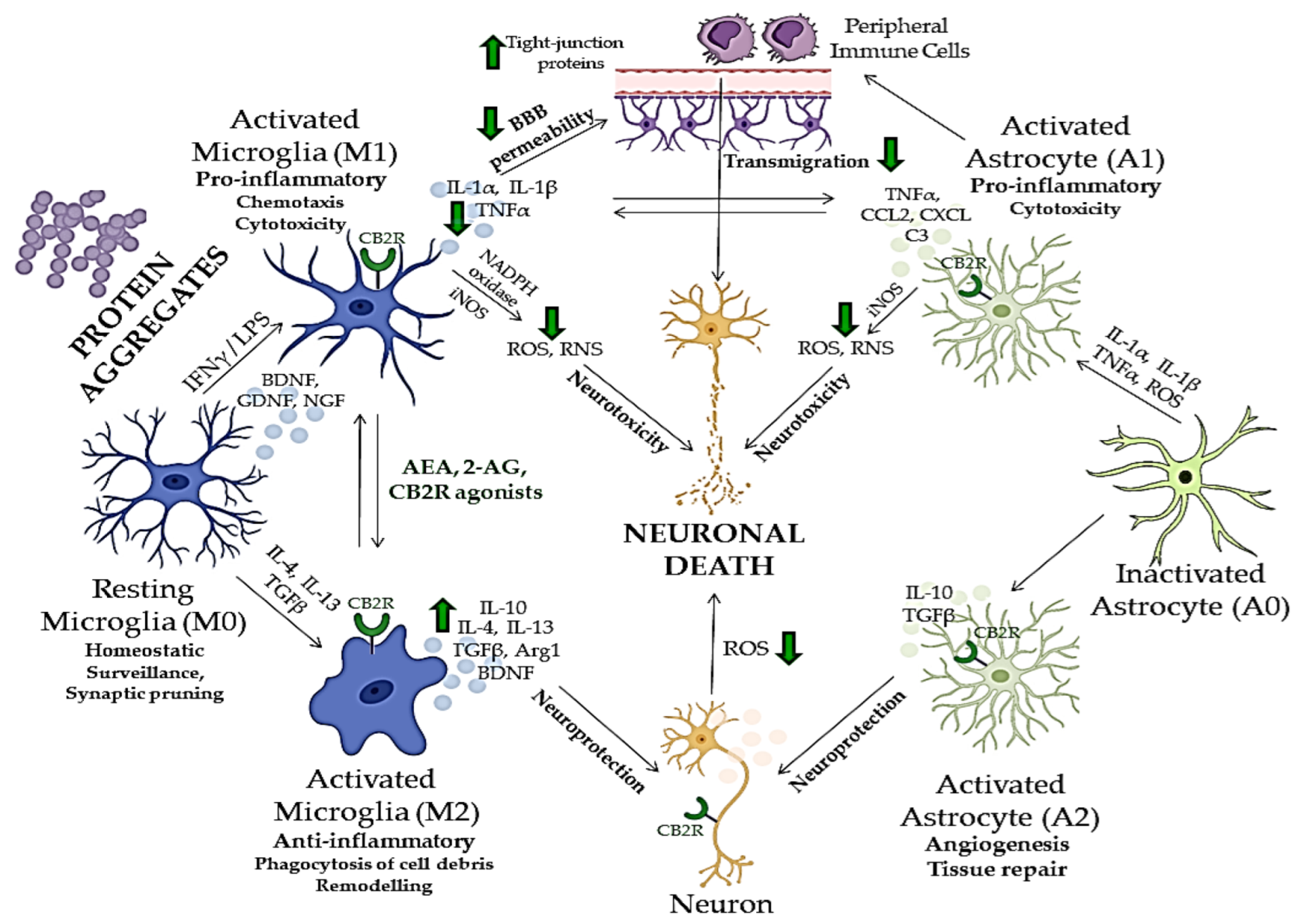
References
- Noor, A.; Zafar, S.; Zerr, I. Neurodegenerative Proteinopathies in the Proteoform Spectrum—Tools and Challenges. Int. J. Mol. Sci. 2021, 22, 1085.
- Knowles, T.P.J.; Vendruscolo, M.; Dobson, C.M. The amyloid state and its association with protein misfolding diseases. Nat. Rev. Mol. Cell Biol. 2014, 15, 384–396.
- Ciccocioppo, F.; Bologna, G.; Ercolino, E.; Pierdomenico, L.; Simeone, P.; Lanuti, P.; Pieragostino, D.; Del Boccio, P.; Marchisio, M.; Miscia, S. Neurodegenerative diseases as proteinopathies-driven immune disorders. Neural Regen. Res. 2020, 15, 850–856.
- Winklhofer, K.F.; Tatzelt, J.; Haass, C. The two faces of protein misfolding: Gain- and loss-of-function in neurodegenerative diseases. EMBO J. 2008, 272, 336–349.
- Sami, N.; Rahman, S.; Kumar, V.; Zaidi, S.; Islam, A.; Ali, S.; Ahmad, F.; Hassan, M.I. Protein aggregation, misfolding and consequential human neurodegenerative diseases. Int. J. Neurosci. 2017, 127, 1047–1057.
- Soto, C.; Pritzkow, S. Protein misfolding, aggregation, and conformational strains in neurodegenerative diseases. Nat. Neurosci. 2018, 21, 1332–1340.
- Chitnis, T.; Weiner, H.L. CNS inflammation and neurodegeneration. J. Clin. Investig. 2017, 127, 3577–3587.
- Pagano, C.; Navarra, G.; Coppola, L.; Avilia, G.; Bifulco, M.; Laezza, C. Cannabinoids: Therapeutic Use in Clinical Practice. Int. J. Mol. Sci. 2022, 236, 3344.
- Kibret, B.G.; Ishiguro, H.; Horiuchi, Y.; Onaivi, E.S. New Insights and Potential Therapeutic Targeting of CB2 Cannabinoid Receptors in CNS Disorders. Int. J. Mol. Sci. 2022, 23, 975.
- García-Gutiérrez, M.S.; Navarrete, F.; Gasparyan, A.; Manzanares, J. Therapeutic potential of the cannabinoid receptor 2 in neuropsychiatry. Explor. Neuroprot. Ther. 2021, 1, 55–71.
- Komorowska-Müller, J.A.; Schmöle, A.C. CB2 Receptor in Microglia: The Guardian of Self-Control. Int. J. Mol. Sci. 2021, 22, 19.
- Sweeney, P.; Park, H.; Baumann, M.; Dunlop, J.; Frydman, J.; Kopito, R.; McCampbell, A.; Leblanc, G.; Venkateswaran, A.; Nurmi, A.; et al. Protein misfolding in neurodegenerative diseases: Implications and strategies. Transl. Neurodegener. 2017, 6, 6.
- Schattling, B.; Engler, J.B.; Volkmann, C.; Rothammer, N.; Woo, M.S.; Petersen, M.; Winkler, I.; Kaufmann, M.; Rosenkranz, S.C.; Fejtova, A.; et al. Bassoon proteinopathy drives neurodegeneration in multiple sclerosis. Nat. Neurosci. 2019, 22, 887–896.
- Hartl, U.F. Protein misfolding diseases. Annu. Rev. Biochem. 2017, 86, 21–26.
- Marsh, A.P. Molecular mechanisms of proteinopathies across neurodegenerative disease: A review. Neurol. Res. Pract. 2019, 1, 35.
- Ciechanover, A.; Kwon, Y.T. Protein quality control by molecular chaperones in neurodegeneration. Front. Neurosci. 2017, 11, 185.
- Hayashida, N.; Fujimoto, M.; Tan, K.; Prakasam, R.; Shinkawa, T.; Li, L.; Ichikawa, H.; Takii, R.; Nakai, A. Heat shock factor 1 ameliorates proteotoxicity in cooperation with the transcription factor NFAT. EMBO J. 2010, 29, 3459–3469.
- Hipp, M.S.; Park, S.H.; Hartl, U.U. Proteostasis impairment in protein-misfolding and aggregation diseases. Trends Cell Biol. 2014, 24, 506–514.
- Nonaka, T.; Hasegawa, M. A cellular model to monitor proteasome dysfunction by α-synuclein. Biochemistry 2009, 48, 8014–8022.
- Finley, D. Recognition and processing of ubiquitin-protein conjugates by the proteasome. Annu. Rev. Biochem. 2009, 78, 477–513.
- Brundin, P.; Melki, R.; Kopito, R. Prion-like transmission of protein aggregates in neurodegenerative diseases. Nat. Rev. Mol. Cell Biol. 2010, 11, 301–307.
- Brettschneider, J.; Del Tredici, K.; Lee, V.M.Y.; Trojanowski, J.Q. Spreading of pathology in neurodegenerative diseases: A focus on human studies. Nat. Rev. Neurosci. 2015, 16, 109–120.
- Galpern, W.R.; Lang, A.E. Interface between tauopathies and synucleinopathies: A tale of two proteins. Ann. Neurol. 2006, 59, 449–458.
- Gidalevitz, T.; Krupinski, T.; Garcia, S.; Morimoto, R.I. Destabilizing protein polymorphisms in the genetic background direct phenotypic expression of mutant SOD1 toxicity. PLoS Genet. 2009, 5, e1000399.
- Wu, J.W.; Hussaini, S.A.; Bastille, I.M.; Rodriguez, G.A.; Mrejeru, A.; Rilett, K.; Sanders, D.W.; Cook, C.; Fu, H.; Boonen, R.A.; et al. Neuronal activity enhances tau propagation and tau pathology in vivo. Nat. Neurosci. 2016, 19, 1085–1092.
- Lee, J.G.; Takahama, S.; Zhang, G.; Tomarev, S.I.; Ye, Y. Unconventional secretion of misfolded proteins promotes adaptation to proteasome dysfunction in mammalian cells. Nat. Cell Biol. 2016, 18, 765–776.
- Shankar, G.M.; Li, S.; Mehta, T.H.; Garcia-Munoz, A.; Shepardson, N.E.; Smith, I.; Brett, F.M.; Farrell, M.A.; Rowan, M.J.; Lemere, C.A.; et al. Amyloid-β protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat. Med. 2008, 14, 837–842.
- Kopeikina, K.J.; Hyman, B.J.; Spires-Jones, T.L. Soluble forms of tau are toxic in Alzheimer’s disease. Transl. Neurosci. 2012, 3, 223–233.
- Ingelsson, M. Alpha-synuclein oligomers-neurotoxic molecules in Parkinson’s disease and other Lewy body disorders. Front. Neurosci. 2016, 10, 408.
- Majd, S.; Power, J.H.; Grantham, H.J.M. Neuronal response in Alzheimer’s and Parkinson’s disease: The effect of toxic proteins on intracellular pathway. BMC Neurosci. 2015, 16, 69.
- Ben Haim, L.; Carrillo-de Sauvage, M.A.; Ceyzériat, K.; Escartin, C. Elusive roles for reactive astrocytes in neurodegenerative diseases. Front. Cell. Neurosci. 2015, 9, 278.
- Jiang, T.; Sun, Q.; Chen, S. Oxidative stress: A major pathogenesis and potential therapeutic target of antioxidative agents in Parkinson’s disease and Alzheimer’s disease. Prog. Neurobiol. 2016, 147, 1–19.
- Angelova, P.R.; Abramov, A.Y. Alpha-synuclein and beta-amyloid—Different targets, same players: Calcium, free radicals and mitochondria in the mechanism of neurodegeneration. Biochem. Biophys. Res. Commun. 2017, 483, 1110–1115.
- Taylor, R.C.; Dillin, A. Aging as an event of proteostasis collapse. Cold Spring Harb. Perspect. Biol. 2011, 3, a004440.
- Höhn, A.; Weber, D.; Jung, T.; Ott, C.; Hugo, M.; Kochlik, B.; Kehm, R.; König, J.; Grune, T.; Castro, J.P. Happily (n)ever after: Aging in the context of oxidative stress, proteostasis loss and cellular senescence. Redox Biol. 2017, 11, 482–501.
- Heppner, F.L.; Ransohoff, R.M.; Becher, B. Immune attack: The role of inflammation in Alzheimer disease. Nat. Rev. Neurosci. 2015, 16, 358–372.
- Chakrabarty, P.; Li, A.; Ceballos-Diaz, C.; Eddy, J.A.; Funk, C.C.; Moore, B.; DiNunno, N.; Rosario, A.M.; Cruz, P.E.; Verbeecket, C.; et al. IL-10 Alters Immunoproteostasis in APP Mice, Increasing Plaque Burden and Worsening Cognitive Behavior. Neuron 2015, 85, 519–533.
- Guillot-Sestier, M.V.; Doty, K.R.; Gate, D.; Rodriguez, J., Jr.; Leung, B.P.; Rezai-Zadeh, K.; Town, T. Il10 deficiency rebalances innate immunity to mitigate Alzheimer-like pathology. Neuron 2015, 85, 534–548.
- Deleidi, M.; Jäggle, M.; Rubino, G. Immune ageing, dysmetabolism and inflammation in neurological diseases. Front. Neurosci. 2015, 9, 172.
- Currais, A.; Fischer, W.; Maher, P.; Schubert, D. Intraneuronal protein aggregation as a trigger for inflammation and neurodegeneration in the aging brain. FASEB J. 2017, 31, 5–10.
- Battista, N.; Di Tommaso, M.; Bari, M.; Maccarrone, M. The endocannabinoid system: An overview. Front. Behav. Neurosci. 2012, 6, 9.
- Aizpurua-Olaizola, O.; Elezgarai, I.; Rico-Barrio, I.; Zarandona, I.; Etxebarria, N.; Usobiaga, A. Targeting the endocannabinoid system: Future therapeutic strategies. Drug Discov. Today 2017, 22, 105–110.
- Lu, H.C.; Mackie, K. Review of the Endocannabinoid System. Biol Psychiatry. Cogn. Neurosci. Neuroimaging 2021, 6, 607–615.
- Chanda, D.; Neumann, D.; Glatz, J.F.C. The endocannabinoid system: Overview of an emerging multi-faceted therapeutic target. Prostaglandins Leukot. Essent. Fat. Acids 2019, 140, 51–56.
- Lu, H.C.; MacKie, K. An introduction to the endogenous cannabinoid system. Biol. Psychiatry 2016, 79, 516–525.
- Glass, M.; Dragunow, M.; Faull, R.L.M. Cannabinoid receptors in the human brain: A detailed anatomical and quantitative autoradiographic study in the fetal, neonatal and adult human brain. Neuroscience 1997, 77, 299–318.
- Biegon, A.; Kerman, I.A. Autoradiographic study of pre- and postnatal distribution of cannabinoid receptors in human brain. Neuroimage 2001, 14, 1463–1468.
- Morris, G.; Walder, K.; Kloiber, S.; Amminger, P.; Berk, M.; Bortolasci, C.C.; Maes, M.; Puri, B.K.; Carvalho, A.F. The endocannabinoidome in neuropsychiatry: Opportunities and potential risks. Pharmacol. Res. 2021, 170, 105729.
- Di Iorio, G.; Lupi, M.; Sarchione, F.; Matarazzo, I.; Santacroce, R.; Petruccelli, F.; Martinotti, G.; Di Giannantonio, M. The Endocannabinoid System: A Putative Role in Neurodegenerative Diseases. Int. J. High Risk Behav. Addict. 2013, 2, 100–106.
- Marco, E.M.; García-Gutiérrez, M.S.; Bermúdez-Silva, F.J.; Moreira, F.A.; Guimarães, F.; Manzanares, J.; Viveros, M.P. Endocannabinoid system and psychiatry: In search of a neurobiological basis for detrimental and potential therapeutic effects. Front. Behav. Neurosci. 2011, 5, 63.
- Galiègue, S.; Mary, S.; Marchand, J.; Dussossoy, D.; Carrière, D.; Carayon, P.; Bouaboula, M.; Shire, D.; Le Fur, G.; Casellas, P. Expression of Central and Peripheral Cannabinoid Receptors in Human Immune Tissues and Leukocyte Subpopulations. Eur. J. Biochem. 1995, 232, 54–61.
- Zou, S.; Kumar, U. Cannabinoid Receptors and the Endocannabinoid System: Signaling and Function in the Central Nervous System. Int. J. Mol. Sci. 2018, 19, 833.
- Howlett, A.C. The cannabinoid receptors. Prostaglandins Other Lipid Mediat. 2002, 68, 619–631.
- Paloczi, J.; Varga, Z.V.; Hasko, G.; Pacher, P. Neuroprotection in Oxidative Stress-Related Neurodegenerative Diseases: Role of Endocannabinoid System Modulation. Antioxid. Redox Signal. 2017, 29, 75–108.
- Tadijan, A.; Vlasic, I.; Vlainic, J.; Dikic, D.; Orsolic, N.; Jazvinscak Jembrek, M. Intracellular Molecular Targets and Signaling Pathways Involved in Antioxidative and Neuroprotective Effects of Cannabinoids in Neurodegenerative Conditions. Antioxidants 2022, 11, 2049.
- Ehrhart, J.; Obregon, D.; Mori, T.; Hou, H.; Sun, N.; Bai, Y.; Klein, T.; Fernandez, F.; Tan, J.; Shytle, R.D. Stimulation of cannabinoid receptor 2 (CB2) suppresses microglial activation. J. Neuroinflamm. 2005, 12, 29–42.
- Ma, L.; Jia, J.; Liu, X.; Bai, F.; Wang, Q.; Xiong, L. Activation of murine microglial N9 cells is attenuated through cannabinoid receptor CB2 signaling. Biochem. Biophys. Res. Commun. 2015, 458, 92–97.
- Cristino, L.; Bisogno, T.; Di Marzo, V. Cannabinoids and the expanded endocannabinoid system in neurological disorders. Nat. Rev. Neurol. 2020, 6, 9–29.
- Pertwee, R.G.; Howlett, A.C.; Abood, M.E.; Alexander, S.P.H.; Di Marzo, V.; Elphick, M.R.; Greasley, P.J.; Hansen, H.S.; Kunos, G.; Mackie, K.; et al. International Union of Basic and Clinical Pharmacology. LXXIX. Cannabinoid receptors and their ligands: Beyond CB1 and CB2. Pharmacol. Rev. 2010, 62, 588–631.
- Joshi, N.; Onaivi, E.S. Endocannabinoid System Components: Overview and Tissue Distribution. In Recent Advances in Cannabinoid Physiology and Pathology; Advances in Experimental Medicine and Biology; Springer: Cham, Switzerland, 2019; Volume 1162, pp. 1–12.
- Bisogno, T.; Berrendero, F.; Ambrosino, G.; Cebeira, M.; Ramos, J.A.; Fernandez-Ruiz, J.J.; Di Marzo, V. Brain regional distribution of endocannabinoids: Implications for their biosynthesis and biological function. Biochem. Biophys. Res. Commun. 1999, 256, 377–380.
- Felder, C.C.; Nielsen, A.; Briley, E.M.; Palkovits, M.; Priller, J.; Axelrod, J.; Nguyen, D.N.; Richardson, J.M.; Riggin, R.M.; Koppel, G.A.; et al. Isolation and measurement of the endogenous cannabinoid receptor agonist, anandamide, in brain and peripheral tissues of human and rat. FEBS Lett. 1996, 393, 231–235.
- Castillo, P.E.; Younts, T.J.; Chávez, A.E.; Hashimotodani, Y. Endocannabinoid Signaling and Synaptic Function. Neuron 2012, 76, 70–81.
- Katona, I.; Freund, T.F. Endocannabinoid signaling as a synaptic circuit breaker in neurological disease. Nat. Med. 2008, 14, 923–930.
- Pacher, P.; Batkai, S.; Kunos, G. The Endocannabinoid System as an Emerging Target of Pharmacotherapy. Pharmacol. Rev. 2006, 58, 389–422.
- Murataeva, N.; Straiker, A.; MacKie, K. Parsing the players: 2-arachidonoylglycerol synthesis and degradation in the CNS. Br. J. Pharmacol. 2014, 171, 1379–1391.
- Jung, K.M.; Sepers, M.; Henstridge, C.M.; Lassalle, O.; Neuhofer, D.; Martin, H.; Ginger, M.; Frick, A.; DiPatrizio, N.V.; Mackie, K.; et al. Uncoupling of the endocannabinoid signalling complex in a mouse model of fragile X syndrome. Nat. Commun. 2012, 3, 1080–1091.
- Kano, M.; Ohno-Shosaku, T.; Hashimotodani, Y.; Uchigashima, M.; Watanabe, M. Endocannabinoid-mediated control of synaptic transmission. Physiol. Rev. 2009, 89, 309–380.
- Glaser, S.T.; Abumrad, N.A.; Fatade, F.; Kaczocha, M.; Studholme, K.M.; Deutsch, D.G. Evidence against the presence of an anandamide transporter. Proc. Natl. Acad. Sci. USA 2003, 100, 4269–4274.
- McFarland, M.J.; Porter, A.C.; Rakhshan, F.R.; Rawat, D.S.; Gibbs, R.A.; Barker, E.L. A role for caveolae/lipid rafts in the uptake and recycling of the endogenous cannabinoid anandamide. J. Biol. Chem. 2004, 279, 41991–41997.
- Huang, H.; McIntosh, A.L.; Martin, G.G.; Landrock, D.; Chung, S.; Landrock, K.K.; Dangott, L.J.; Li, S.; Kier, A.B.; Schroeder, F. FABP1: A Novel Hepatic Endocannabinoid and Cannabinoid Binding Protein. Biochemistry 2016, 55, 5243–5255.
- Blankman, J.L.; Simon, G.M.; Cravatt, B.F. A Comprehensive Profile of Brain Enzymes that Hydrolyze the Endocannabinoid 2-Arachidonoylglycerol. Chem. Biol. 2007, 14, 1347–1356.
- Berry, A.J.; Zubko, O.; Reeves, S.J.; Howard, R.J. Endocannabinoid system alterations in Alzheimer’s disease: A systematic review of human studies. Brain Res. 2020, 1749, 147135–147148.
- Behl, T.; Kaur, G.; Bungau, S.; Jhanji, R.; Kumar, A.; Mehta, V.; Zengin, G.; Brata, R.; Hassan, S.S.U.; Fratila, O. Distinctive evidence involved in the role of endocannabinoid signalling in parkinson’s disease: A perspective on associated therapeutic interventions. Int. J. Mol. Sci. 2020, 21, 6235.
- Reddy, V.; Grogan, D.; Ahluwalia, M.; Salles, É.L.; Ahluwalia, P.; Khodadadi, H.; Alverson, K.; Nguyen, A.; Raju, S.P.; Gaur, P.; et al. Targeting the endocannabinoid system: A predictive, preventive, and personalized medicine-directed approach to the management of brain pathologies. EPMA J. 2020, 11, 217–250.
- Jones, É.; Vlachou, S. A Critical Review of the Role of the Cannabinoid Compounds Δ9-Tetrahydrocannabinol (Δ9-THC) and Cannabidiol (CBD) and their Combination in Multiple Sclerosis Treatment. Molecules 2020, 25, 4930.
- Fakhoury, M. Role of the Endocannabinoid System in the Pathophysiology of Schizophrenia. Mol. Neurobiol. 2017, 54, 768–778.
- Papagianni, E.P.; Stevenson, C.W. Cannabinoid Regulation of Fear and Anxiety: An Update. Curr. Psychiatry Rep. 2019, 21, 38–48.
- Huang, W.J.; Chen, W.W.; Zhang, X. Endocannabinoid system: Role in depression, reward and pain control (Review). Mol. Med. Rep. 2016, 14, 2899–2903.
- Davis, J.; Moylan, S.; Harvey, B.H.; Maes, M.; Berk, M. Neuroprogression in schizophrenia: Pathways underpinning clinical staging and therapeutic corollaries. Aust. N. Z. J. Psychiatry 2014, 48, 512–529.
- Berk, M.; Kapczinski, F.; Andreazza, A.C.; Dean, O.M.; Giorlando, F.; Maes, M.; Yücel, M.; Gama, C.S.; Dodd, S.; Dean, B.; et al. Pathways underlying neuroprogression in bipolar disorder: Focus on inflammation, oxidative stress and neurotrophic factors. Neurosci. Biobehav. Rev. 2011, 35, 804–817.
- Di Marzo, V. Targeting the endocannabinoid system: To enhance or reduce? Nat. Rev. Drug Discov. 2008, 7, 438–455.
- Hanuš, L.O.; Meyer, S.M.; Muñoz, E.; Taglialatela-Scafati, O.; Appendino, G. Phytocannabinoids: A unified critical inventory. Nat. Prod. Rep. 2016, 33, 1357–1392.
- Brown, J.D.; Rivera Rivera, K.J.; Hernandez, L.Y.C.; Doenges, M.R.; Auchey, I.; Pham, T.; Goodin, A.J. Natural and Synthetic Cannabinoids: Pharmacology, Uses, Adverse Drug Events, and Drug Interactions. J. Clin. Pharmacol. 2021, 61, S37–S52.
- Bie, B.; Wu, J.; Foss, J.F.; Naguib, M. An overview of the cannabinoid type 2 receptor system and its therapeutic potential. Curr. Opin. Anaesthesiol. 2018, 31, 407–414.
- Ruhaak, L.R.; Felth, J.; Karlsson, P.C.; Rafter, J.J.; Verpoorte, R.; Bohlin, L. Evaluation of the Cyclooxygenase Inhibiting Effects of Six Major Cannabinoids Isolated from Cannabis sativa. Biol. Pharm. Bull. 2011, 34, 774–778.
- Pisanti, S.; Malfitano, A.M.; Ciaglia, E.; Lamberti, A.; Ranieri, R.; Cuomo, G.; Abate, M.; Faggiana, G.; Proto, M.C.; Fiore, D.; et al. Cannabidiol: State of the art and new challenges for therapeutic applications. Pharmacol. Ther. 2017, 175, 133–150.
- Thakur, G.A.; Duclos, R.I., Jr.; Makriyannis, A. Natural cannabinoids: Templates for drug discovery. Life Sci. 2005, 22, 454–466.
- Morales, P.; Hurst, D.P.; Reggio, P.H. Molecular Targets of the Phytocannabinoids: A Complex Picture. Prog. Chem. Org. Nat. Prod. 2017, 103, 103–131.
- Schlatter, J.; Atta, U.-R. Synthetic Cannabinoids: Synthesis and Biological Activities. In Studies in Natural Products Chemistry; Elsevier: Amsterdam, The Netherlands, 2014; pp. 291–311.
- Abate, G.; Uberti, D.; Tambaro, S. Potential and Limits of Cannabinoids in Alzheimer’s Disease Therapy. Biology 2021, 10, 542.
- Fernández-Ruiz, M.; Romero, J.; Velasco, G.; Tolon, R.M.; Ramos, J.A.; Guzman, M. Cannabinoid CB2 receptor: A new target for controlling neural cell survival? Trends Pharmacol. Sci. 2007, 28, 39–45.
- Munro, S.; Thomas, K.L.; Abu-Shaar, M. Molecular characterization of a peripheral receptor for cannabinoids. Nature 1993, 365, 61–65.
- Schatz, A.R.; Lee, M.; Condie, R.B.; Pulaski, J.T.; Kaminski, N.E. Cannabinoid receptors CB1 and CB2: A characterization of expression and adenylate cyclase modulation within the immune system. Toxicol. Appl. Pharmacol. 1997, 142, 278–287.
- Polini, B.; Cervetto, C.; Carpi, S.; Pelassa, S.; Gado, F.; Ferrisi, R.; Bertini, S.; Nieri, P.; Marcoli, M.; Manera, C. Positive Allosteric Modulation of CB1 and CB2 Cannabinoid Receptors Enhances the Neuroprotective Activity of a Dual CB1R / CB2R Orthosteric Agonist. Life 2020, 1, 333.
- Brown, S.M.; Wager-Miller, J.; Mackie, K. Cloning and Molecular Characterization of the Rat CB2 Cannabinoid Receptor. Biochim. Biophys. Acta 2002, 1576, 255–264.
- Chen, D.J.; Gao, M.; Gao, F.F.; Su, Q.X.; Wu, J. Brain cannabinoid receptor 2: Expression, function and modulation. Acta Pharmacol. Sin. 2017, 38, 312–316.
- Onaivi, E.S.; Ishiguro, H.; Gu, S.; Liu, Q.R. CNS effects of CB2 cannabinoid receptors: Beyond neuro-immuno-cannabinoid activity. J. Psychopharmacol. 2012, 26, 92–103.
- Buckley, N.E.; McCoy, K.L.; Mezey, E.; Bonner, T.; Zimmer, A.; Felder, C.C.; Glass, M.; Zimmer, A. Immunomodulation by cannabinoids is absent in mice deficient for the cannabinoid CB2 receptor. Eur. J. Pharmacol. 2000, 396, 141–149.
- Gong, J.P.; Onaivi, E.S.; Ishiguro, H.; Liu, Q.R.; Tagliaferro, P.A.; Brusco, A.; Uhl, G.R. Cannabinoid CB2 receptors: Immunohistochemical localization in rat brain. Brain Res. 2006, 1071, 10–23.
- Onaivi, E.S.; Ishiguro, H.; Gong, J.P.; Patel, S.; Meozzi, P.A.; Myers, L.; Perchuk, A.; Mora, Z.; Tagliaferro, P.A.; Gardner, E.; et al. Functional expression of brain neuronal CB2 cannabinoid receptors are involved in the effects of drugs of abuse and in depression. Ann. N. Y. Acad. Sci. 2008, 1139, 434–449.
- Wang, M.; Liu, H.; Ma, Z. Roles of the Cannabinoid System in the Basal Ganglia in Parkinson’s Disease. Front. Cell. Neurosci. 2022, 16, 832854.
- Di Marzo, V. New approaches and challenges to targeting the endocannabinoid system. Nat. Rev. Drug Discov. 2018, 17, 623–639.
- Brusco, A.; Tagliaferro, P.; Saez, T.; Onaivi, E.S. Postsynaptic localization of CB2 cannabinoid receptors in the rat hippocampus. Synapse 2008, 62, 944–949.
- Onaivi, E.S.; Ishiguro, H.; Gong, J.P.; Patel, S.; Perchuk, A.; Meozzi, P.A.; Myers, L.; Mora, Z.; Tagliaferro, P.; Gardner, E.; et al. Discovery of the presence and functional expression of cannabinoid CB2 receptors in brain. Ann. N. Y. Acad. Sci. 2006, 1074, 514–536.
- Chin, C.L.; Tovcimak, A.E.; Hradil, V.P.; Seifert, T.R.; Hollingsworth, P.R.; Chandran, P.; Zhu, C.Z.; Gauvin, D.; Pai, M.; Wetter, J.; et al. Differential effects of cannabinoid receptor agonists on regional brain activity using pharmacological MRI. Br. J. Pharmacol. 2008, 153, 367–379.
- Li, Y.; Kim, J. Neuronal Expression of CB2 Cannabinoid Receptor MRNAs in the Mouse Hippocampus. Neuroscience 2015, 311, 253–267.
- Vaseghi, S.; Nasehi, M.; Zarrindast, M.R. How Do Stupendous Cannabinoids Modulate Memory Processing via Affecting Neurotransmitter Systems? Neurosci. Biobehav. Rev. 2021, 120, 173–221.
- Zhang, H.Y.; Gao, M.; Liu, Q.R.; Bi, G.H.; Li, X.; Yang, H.J.; Gardner, E.L.; Wu, J.; Xi, Z.X. Cannabinoid CB2 Receptors Modulate Midbrain Dopamine Neuronal Activity and Dopamine-Related Behavior in Mice. Proc. Natl. Acad. Sci. USA 2014, 111, E5007–E5015.
- Eljaschewitsch, E.; Witting, A.; Mawrin, C.; Lee, T.; Schmidt, P.M.; Wolf, S.; Hoertnagl, H.; Raine, C.S.; Schneider-Stock, R.; Nitsch, R.; et al. The Endocannabinoid Anandamide Protects Neurons during CNS Inflammation by Induction of MKP-1 in Microglial Cells. Neuron 2006, 49, 67–79.
- Navarro, G.; Morales, P.; Rodríguez-Cueto, C. Targeting Cannabinoid CB2 Receptors in the Central Nervous System. Medicinal Chemistry Approaches with Focus on Neurodegenerative Disorders. Front. Neurosci. 2016, 10, 406.
- Malek, N.; Popiolek-Barczyk, K.; Mika, J.; Przewlocka, B.; Starowicz, K. Anandamide, Acting via CB2 Receptors, Alleviates LPS-Induced Neuroinflammation in Rat Primary Microglial Cultures. Neural Plast. 2015, 2015, 13069.
- Aghazadeh Tabrizi, M.; Baraldi, P.G.; Borea, P.A.; Varani, K. Medicinal Chemistry, Pharmacology, and Potential Therapeutic Benefits of Cannabinoid CB2Receptor Agonists. Chem. Rev. 2016, 116, 519–560.
- Reiner, A.; Heldt, S.A.; Presley, C.S.; Guley, N.H.; Elberger, A.J.; Deng, Y.; D’Surney, L.; Rogers, J.T.; Ferrell, J.; Bu, W.; et al. Motor, Visual and Emotional Deficits in Mice after Closed-Head Mild Traumatic Brain Injury Are Alleviated by the Novel CB2 Inverse Agonist SMM-189. Int. Mol. J. Sci. 2015, 16, 758–787.
- Presley, C.S.; Mustafa, S.M.; Abidi, A.H.; Moore, B.M. Synthesis and biological evaluation of (3′,5′-dichloro-2,6-dihydroxy-biphenyl-4-yl)-aryl/alkyl-methanone selective CB2 inverse agonist. Bioorg. Med. Chem. 2015, 23, 5390–5401.
- Chicca, A.; Gachet, M.S.; Petrucci, V.; Schuehly, W.; Charles, R. 4′-O-methylhonokiol increases levels of 2-arachidonoyl glycerol in mouse brain via selective inhibition of its COX-2-mediated oxygenation. J. Neuroinflamm. 2015, 12, 89–105.
- Nimczick, M.; Decker, M. New Approaches in the Design and Development of Cannabinoid Receptor Ligands: Multifunctional and Bivalent Compounds. ChemMedChem 2015, 10, 773–786.
- Nimczick, M.; Pemp, D.; Darras, F.H.; Chen, X.; Heilmann, J.; Decker, M. Synthesis and biological evaluation of bivalent cannabinoid receptor ligands based on hCB2R selective benzimidazoles reveal unexpected intrinsic properties. Bioorg. Med. Chem. 2014, 22, 3938–3946.
- Khajehali, E.; Malone, D.T.; Glass, M.; Sexton, P.M.; Christopoulos, A.; Leach, K. Biased Agonism and Biased Allosteric Modulation at the CB 1 Cannabinoid Receptors. Mol. Pharmacol. 2015, 88, 368–379.
- Dhopeshwarkar, A.; Mackie, K. CB2 cannabinoid receptors as a therapeutic target-what does the future hold? Mol. Pharmacol. 2014, 86, 430–437.
- Morales, P.; Goya, P.; Jagerovic, N.; Hernandez-Folgado, L. Allosteric Modulators of the CB1 Cannabinoid Receptor: A Structural Update Review. Cannabis Cannabionid Res. 2016, 1, 22–30.
- Saxena, S.; Caroni, P. Selective Neuronal Vulnerability in Neurodegenerative Diseases: From Stressor Thresholds to Degeneration. Neuron 2011, 71, 35–48.
- Centonze, D.; Finazzi-Agrò, A.; Bernardi, G.; Maccarrone, M. The Endocannabinoid System in Targeting Inflammatory Neurodegenerative Diseases. Trends Pharmacol. Sci. 2007, 28, 180–187.
- Kovacs, G.G.; Adle-Biassette, H.; Milenkovic, I.; Cipriani, S.; Van Scheppingen, J.; Aronica, E. Linking pathways in the developing and aging brain with neurodegeneration. Neuroscience 2014, 269, 152–172.
- Di Marzo, V.; Stella, N.; Zimmer, A. Endocannabinoid signalling and the deteriorating brain. Nat. Rev. Neurosci. 2015, 16, 30–42.
- Scotter, E.L.; Abood, M.E.; Glass, M. The Endocannabinoid System as a Target for the Treatment of Neurodegenerative Disease. Br. J. Pharmacol. 2010, 160, 480–498.
- Tao, R.; Li, C.; Jaffe, A.E.; Shin, J.H.; Deep-Soboslay, A.; Yamin, R.; Weinberger, D.R.; Hyde, T.M.; Kleinman, J.E. Cannabinoid receptor CNR1 expression and DNA methylation in human prefrontal cortex, hippocampus and caudate in brain development and schizophrenia. Transl. Psychiatry 2020, 10, 158–171.
- Monteleone, P.; Bifulco, M.; Maina, G.; Tortorella, A.; Gazzerro, P.; Proto, M.C.; Di Filippo, C.; Monteleone, F.; Canestrelli, B.; Buonerba, G.; et al. Investigation of CNR1 and FAAH endocannabinoid gene polymorphisms in bipolar disorder and major depression. Pharmacol. Res. 2010, 61, 400–404.
- Demers, C.H.; Drabant Conley, E.; Bogdan, R.; Hariri, A.R. Interactions Between Anandamide and Corticotropin-Releasing Factor Signaling Modulate Human Amygdala Function and Risk for Anxiety Disorders: An Imaging Genetics Strategy for Modeling Molecular Interactions. Biol. Psychiatry 2016, 80, 356–362.
- Nguyen, T.; Thomas, B.F.; Zhang, Y. Overcoming the Psychiatric Side Effects of the Cannabinoid CB1 Receptor Antagonists: Current Approaches for Therapeutics Development. Curr. Top. Med. Chem. 2019, 19, 1418–1435.
- Moreira, F.A.; Grieb, M.; Lutz, B. Central side-effects of therapies based on CB1 cannabinoid receptor agonists and antagonists: Focus on anxiety and depression. Best Pract. Res. Clin. Endocrinol. Metab. 2009, 23, 133–144.
- Emadi, L.; Jonaidi, H.; Amir Abad, E.H. The role of central CB2 cannabinoid receptors on food intake in neonatal chicks. J. Comp. Physiol. A Neuroethol. Sens. Neural Behav. Physiol. 2011, 197, 1143–1147.
- Liu, Q.R.; Canseco-Alba, A.; Zhang, H.Y.; Tagliaferro, P.; Chung, M.; Dennis, E.; Sanabria, B.; Schanz, N.; Escosteguy-Neto, J.C.; Ishiguro, H.; et al. Cannabinoid type 2 receptors in dopamine neurons inhibits psychomotor behaviors, alters anxiety, depression and alcohol preference. Sci. Rep. 2017, 7, 17410.
- García-Gutiérrez, M.S.; Manzanares, J. Overexpression of CB2 cannabinoid receptors decreased vulnerability to anxiety and impaired anxiolytic action of alprazolam in mice. J. Psychopharmacol. 2011, 25, 111–120.
- Geresu, B.; Canseco-Alba, A.; Sanabria, B.; Lin, Z.; Liu, Q.R.; Onaivi, E.S. Involvement of CB2 receptors in the neurobehavioral effects of Catha edulis (Vahl) Endl. (khat) in mice. Molecules 2019, 24, 3164.
- Svíženská, I.H.; Brázda, V.; Klusáková, I.; Dubový, P. Bilateral Changes of Cannabinoid Receptor Type 2 Protein and mRNA in the Dorsal Root Ganglia of a Rat Neuropathic Pain Model. J. Histochem. Cytochem. 2013, 61, 529–547.
- Yu, S.J.; Reiner, D.; Shen, H.; Wu, K.J.; Liu, Q.R.; Wang, Y. Time-dependent protection of CB2 receptor agonist in stroke. PLoS ONE 2015, 10, e0132487.
- Ishiguro, H.; Horiuchi, Y.; Ishikawa, M.; Koga, M.; Imai, K.; Suzuki, Y.; Morikawa, M.; Inada, T.; Watanabe, Y.; Takahashi, M.; et al. Brain Cannabinoid CB2 Receptor in Schizophrenia. Biol. Psychiatry 2010, 67, 974–982.
- Ortega-Alvaro, A.; Aracil-Fernández, A.; García-Gutiérrez, M.S.; Navarrete, F.; Manzanares, J. Deletion of CB2 Cannabinoid Receptor Induces Schizophrenia-Related Behaviors in Mice. Neuropsychopharmacology 2011, 36, 1489–1504.
- Lopez-Rodriguez, A.B.; Acaz-Fonseca, E.; Viveros, M.P.; Garcia-Segura, L.M. Changes in cannabinoid receptors, aquaporin 4 and vimentin expression after traumatic brain injury in adolescent male mice. Association with edema and neurological deficit. PLoS ONE 2015, 10, e0128782.
- Romero-Sandoval, E.A.; Horvath, R.; Landry, R.P.; DeLeo, J.A. Cannabinoid receptor type 2 activation induces a microglial anti-inflammatory phenotype and reduces migration via MKP induction and ERK dephosphorylation. Mol. Pain 2009, 5, 25.
- Agudelo, M.; Yndart, A.; Morrison, M.; Figueroa, G.; Muñoz, K.; Samikkannu, T.; Nair, M.P. Differential Expression and Functional Role of Cannabinoid Genes in Alcohol Users. Drug Alcohol Depend. 2013, 133, 789–793.
- Geresu, B.; Engidawork, E. Catha edulis F. (Khat) Reverses Haloperidol but Not Morphine Induced Motor Deficits Following Acute and Subacute Administration in Mice. Ethiop. Pharm. J. 2012, 28, 117–130.
- Concannon, R.M.; Okine, B.N.; Finn, D.P.; Dowd, E. Differential Upregulation of the Cannabinoid CB2 Receptor in Neurotoxic and Inflammation-Driven Rat Models of Parkinson’s Disease. Exp. Neurol. 2015, 269, 133–141.
- Solas, M.; Francis, P.T.; Franco, R.; Ramirez, M.J. CB2 Receptor and Amyloid Pathology in Frontal Cortex of Alzheimer’s Disease Patients. Neurobiol. Aging 2013, 34, 805–808.
- Facchinetti, F.; Del Giudice, E.; Furegato, S.; Passarotto, M.; Leon, A. Cannabinoids ablate release of TNFα in rat microglial cells stimulated with lypopolysaccharide. Glia 2003, 41, 161–168.
- Lin, L.; Yihao, T.; Zhou, F.; Yin, N.; Qiang, T.; Haowen, Z.; Qianwei, C.; Jun, T.; Yuan, Z.; Gang, Z.; et al. Inflammatory regulation by driving microglial M2 polarization: Neuroprotective effects of cannabinoid receptor-2 activation in intracerebral hemorrhage. Front. Immunol. 2017, 8, 112.
- Ishiguro, H.; Kibret, B.G.; Horiuchi, Y.; Onaivi, E.S. Potential Role of Cannabinoid Type 2 Receptors in Neuropsychiatric and Neurodegenerative Disorders. Front. Psychiatry 2022, 13, 828895.
- McIntosh, T.K.; Smith, D.H.; Meaney, D.F.; Kotapka, M.J.; Gennarelli, T.A.; Graham, D.I. Neuropathological Sequelae of Traumatic Brain Injury: Relationship to Neurochemical and Biomechanical Mechanisms. Lab. Investig. 1996, 74, 315–342.
- Block, M.L.; Hong, J.S. Microglia and Inflammation-Mediated Neurodegeneration: Multiple Triggers with a Common Mechanism. Prog. Neurobiol. 2005, 76, 77–98.
- Kreutzberg, G.W. Microglia: A Sensor for Pathological Events in the CNS. Trends Neurosci. 1996, 19, 312–318.
- Kurkowska-Jastrzȩbska, I.; Wrońska, A.; Kohutnicka, M.; Członkowski, A.; Członkowska, A. MHC Class II Positive Microglia and Lymphocytic Infiltration Are Present in the Substantia Nigra and Striatum in Mouse Model of Parkinson’s Disease. Acta Neurobiol. Exp. 1999, 59, 1–8.
- Vasincu, A.; Rusu, R.N.; Ababei, D.C.; Larion, M.; Bild, W.; Stanciu, G.D.; Solcan, C.; Bild, V. Endocannabinoid Modulation in Neurodegenerative Diseases: In Pursuit of Certainty. Biology 2022, 11, 440.
- Götz, J.; Ittner, L.M.; Schonrock, N. Alzheimer’s Disease and Frontotemporal Dementia: Prospects of a Tailored Therapy? Med. J. Aust. 2006, 185, 381–384.
- Spillantini, M.G.; Crowther, R.A.; Jakes, R.; Hasegawa, M.; Goedert, M. α-Synuclein in Filamentous Inclusions of Lewy Bodies from Parkinson’s Disease and Dementia with Lewy Bodies. Proc. Natl. Acad. Sci. USA 1998, 95, 6469–6473.
- Benkler, C.; O’Neil, A.L.; Slepian, S.; Qian, F.; Weinreb, P.H.; Rubin, L.L. Aggregated SOD1 Causes Selective Death of Cultured Human Motor Neurons. Sci. Rep. 2018, 8, 16393.
- Hoffner, G.; Souès, S.; Djian, P. Aggregation of Expanded Huntingtin in the Brains of Patients with Huntington Disease. Prion 2007, 1, 26.
- Hickman, S.; Izzy, S.; Sen, P.; Morsett, L.; El Khoury, J. Microglia in Neurodegeneration. Nat. Neurosci. 2018, 21, 1359.
- Panatier, A.; Robitaille, R. The Soothing Touch: Microglial Contact Influences Neuronal Excitability. Dev. Cell 2012, 23, 1125–1126.
- Nimmerjahn, A.; Kirchhoff, F.; Helmchen, F. Neuroscience: Resting Microglial Cells Are Highly Dynamic Surveillants of Brain Parenchyma In Vivo. Science 2005, 308, 1314–1318.
- Paolicelli, R.C.; Bolasco, G.; Pagani, F.; Maggi, L.; Scianni, M.; Panzanelli, P.; Giustetto, M.; Ferreira, T.A.; Guiducci, E.; Dumas, L.; et al. Synaptic Pruning by Microglia Is Necessary for Normal Brain Development. Science 2011, 333, 1456–1458.
- Lan, X.; Han, X.; Li, Q.; Yang, Q.W.; Wang, J. Modulators of microglial activation and polarization after intracerebral haemorrhage. Nat. Rev. Neurol. 2017, 13, 420–433.
- Sierra, A.; Abiega, O.; Shahraz, A.; Neumann, H. Janus-Faced Microglia: Beneficial and Detrimental Consequences of Microglial Phagocytosis. Front. Cell. Neurosci. 2013, 7, 6.
- Helmut, K.; Hanisch, U.K.; Noda, M.; Verkhratsky, A. Physiology of Microglia. Physiol. Rev. 2011, 91, 461–553.
- Tang, Y.; Le, W. Differential Roles of M1 and M2 Microglia in Neurodegenerative Diseases. Mol. Neurobiol. 2016, 53, 1181–1194.
- Naguib, M.; Xu, J.J.; Diaz, P.; Brown, D.L.; Cogdell, D.; Bie, B.; Hu, J.; Craig, S.; Hittelman, W.N. Prevention of paclitaxel-induced neuropathy through activation of the central cannabinoid type 2 receptor system. Anesth. Analg. 2012, 114, 1104–1120.
- Tsuda, M.; Shigemoto-Mogami, Y.; Koizumi, S.; Mizokoshi, A.; Kohsaka, S.; Salter, M.W.; Inoue, K. P2X4 receptors induced in spinal microglia gate tactile allodynia after nerve injury. Nature 2003, 424, 778–783.
- Cassano, T.; Calcagnini, S.; Pace, L.; Marco, F.; De Romano, A.; Gaetani, S. Cannabinoid receptor 2 signaling in neurodegenerative disorders: From pathogenesis to a promising therapeutic target. Front. Neurosci. 2017, 11, 30.
- Mecha, M.; Carrillo-Salinas, F.J.; Feliú, A.; Mestre, L.; Guaza, C. Microglia Activation States and Cannabinoid System: Therapeutic Implications. Pharmacol. Ther. 2016, 166, 40–55.
- Turcotte, C.; Blanchet, M.R.; Laviolette, M.; Flamand, N. The CB2 Receptor and Its Role as a Regulator of Inflammation. Cell. Mol. Life Sci. 2016, 73, 4449–4470.
- Correa, F.; Hernangómez, M.; Mestre, L.; Loría, F.; Spagnolo, A.; Docagne, F.; Di Marzo, V.; Guaza, C. Anandamide Enhances IL-10 Production in Activated Microglia by Targeting CB2 Receptors: Roles of ERK1/2, JNK, and NF-ΚB. Glia 2010, 58, 135–147.
- Oddi, S.; Latini, L.; Viscomi, M.T.; Bisicchia, E.; Molinari, M.; Maccarrone, M. Distinct Regulation of NNOS and INOS by CB2 Receptor in Remote Delayed Neurodegeneration. J. Mol. Med. 2012, 90, 371–387.
- Viscomi, M.T.; Oddi, S.; Latini, L.; Pasquariello, N.; Florenzano, F.; Bernardi, G.; Molinari, M.; Maccarrone, M. Selective CB2 Receptor Agonism Protects Central Neurons from Remote Axotomy-Induced Apoptosis through the PI3K/Akt Pathway. J. Neurosci. 2009, 29, 4564–4570.
- Molina-Holgado, F.; Pinteaux, E.; Moore, J.D.; Molina-Holgado, E.; Guaza, C.; Gibson, R.M.; Rothwell, N.J. Endogenous Interleukin-1 Receptor Antagonist Mediates Anti-Inflammatory and Neuroprotective Actions of Cannabinoids in Neurons and Glia. J. Neurosci. 2003, 23, 6470–6474.
- Smith, S.R.; Terminelli, C.; Denhardt, G. Effects of Cannabinoid Receptor Agonist and Antagonist Ligands on Production of Inflammatory Cytokines and Anti-Inflammatory Interleukin-10 in Endotoxemic Mice. J. Pharmacol. Exp. Ther. 2000, 293, 136–150.
- Amenta, P.S.; Jallo, J.I.; Tuma, R.F.; Elliott, M.B. A Cannabinoid Type 2 Receptor Agonist Attenuates Blood-Brain Barrier Damage and Neurodegeneration in a Murine Model of Traumatic Brain Injury. J. Neurosci. Res. 2012, 90, 2293–2305.
- Bachiller, S.; Jiménez-Ferrer, I.; Paulus, A.; Yang, Y.; Swanberg, M.; Deierborg, T.; Boza-Serrano, A. Microglia in Neurological Diseases: A Road Map to Brain-Disease Dependent-Inflammatory Response. Front. Cell. Neurosci. 2018, 12, 488.