Some of the most prevalent neurodegenerative disorders, including Alzheimer’s and Parkinson’s disease, are proteinopathies characterized by the accumulation of specific protein aggregates in the brain. Such misfolded protein aggregates can trigger modulation of the innate and adaptive immune systems and subsequently lead to chronic neuroinflammation that drives the onset and progression of neurodegenerative diseases. Since there is still no effective disease-modifying treatment, new therapeutic targets for neurodegenerative proteinopathies have been sought. The endocannabinoid system, and in particular the cannabinoid CB2 receptors, have been extensively studied, due to their important role in neuroinflammation, especially in microglial cells.
- neurodegenerative diseases
- proteinopathies
- endocannabinoid system
- cannabinoid CB2 receptors
1. Introduction
2. Neurodegenerative Proteinopathies
Neurodegenerative proteinopathies is an umbrella term for neurodegenerative disorders characterized by the formation of misfolded protein aggregates that cause cellular toxicity and contribute to cellular proteostatic collapse [12][13]. According to the pathophysiological hypothesis of neurodegenerative disorders, some proteins change their conformations, consequently gaining neurotoxic activity or losing their normal function by forming small oligomeric or large fibrillar aggregates, leading to neurodegeneration [3]. Neurodegenerative proteinopathies include some of the most common neurodegenerative disorders, such as Alzheimer’s and Parkinson’s disease, as well as Huntington’s disease, multiple sclerosis, amyotrophic lateral sclerosis, etc. The recent findings showing that bassoon proteinopathy drives neurodegeneration in mice and patients with multiple sclerosis are reminiscent of disease pathways in neurodegenerative proteinopathies [13][12]. The neuronal proteome consists of 10,000 to 20,000 different proteins that, in order to fulfill their biological function, must fold in accordance with the instructions encoded in the amino acid sequence [14]. Therefore, to maintain cellular integrity and health, the process of protein folding and its degradation must be well-regulated [15]. However, their biologically active conformation (the native state) is often marginally stable under normal physiological conditions, and even a small polypeptide of ~100 amino acids can adopt many conformations (~1030) under different conditions [14]. It is, therefore, hardly surprising that the process of protein folding is error-prone, leading to misfolded states and off-pathway aggregates [14]. Due to this susceptibility, cells face a continuous stream of misfolded and aggregated proteins (Table 1) and require supportive molecular chaperones called heat shock proteins (Hsp) to refold, degrade, and eliminate them to maintain proteome homeostasis [12][13].| Neurodegenerative Proteinopathy | Misfolded and Aggregated Protein(S) | ||
|---|---|---|---|
| Alzheimer’s disease (AD) | Amyloid beta (Aβ) peptide, tau | ||
| Parkinson’s disease (PD) | α-synuclein | ||
| Huntington´s disease (HD) | Mutant huntingtin (mHtt) | ||
| Multiple sclerosis (MS) | Bassoon presynaptic cytomatrix protein | ||
| Amyotrophic lateral sclerosis (ALS) | Mutant superoxide dismutase 1 (mSOD1), TAR DNA-binding protein 43 (TDP-43) and fused-in-sarcoma (FUS) protein | ||
| Dementia with Lewy bodies (DLB) | α-synuclein | ||
| Frontotemporal lobar degeneration (FTLD) | FTLD-tau, FTLD-TDP, FTLD-FUS | ||
| Creutzfeldt–Jakob disease (CJD) | Protease-resistant cellular prion protein (PrP | Sc | ) |
3. Endocannabinoid System (ECS)
The ECS plays an important role in both the CNS and peripheral nervous system by modulating the neuronal network function and activity [9]. It is a complex molecular system involved in various biological processes such as maintenance of homeostasis, neurogenesis, neuroprotection, cognition, pain, inflammation, learning and memory, as well as pre- and postnatal development [41][42][41,42]. The ECS consists of endogenous cannabinoids (endocannabinoids), cannabinoid receptors (CBR), enzymes and other different proteins important for the transport and metabolism of endocannabinoids (Figure 1) [34][43][34,43]. Endocannabinoids are endogenous signaling lipid mediators that activate CBR and mimic the actions of Δ9-tetrahydrocannabinol (THC) [43]. The biological effects of endocannabinoids are mediated by two members of the large family of G-protein-coupled receptors (GPCR); CB1R and CB2R (Figure 1) [29][44][29,44].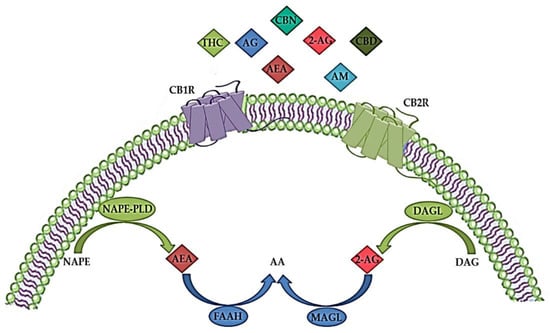
4. Modulation of Cannabinoid Receptor 2 (CB2R)
Marijuana or cannabis (Cannabis sativa) contains about 500 compounds, of which at least 100 are classified as phytocannabinoids with different chemical structures and pharmacological properties, the most abundant natural cannabinoids being THC, cannabidiol (CBD) and cannabinol (CBN) [83]. CBR1 and CRBR2 were first studied as targets of THC in the human brain [84]. THC interacts with both CBR, as an agonist at CB1R and as a weak antagonist at CB2R [85], but also possibly by inhibiting COX enzymes and as an inducer of COX-2 with prolonged exposure [86]. In addition, another natural cannabinoid CBD, which has a low affinity for CBR, may be a CB2R inverse agonist with anti-inflammatory effects [87]. Subsequently, the identification of CBR in the brain suggested the presence of endogenous ligands, and the most studied and characterized endocannabinoids are AEA and 2-AG, which have an affinity for both CBR [84][88][84,88]. It was found that 2-AG acts as a full agonist, while AEA acts as a weak partial agonist for both CB1R and CB2R [85]. CP 55,940 was the first synthetic cannabinoid analog to be synthesized, followed by several others. Although many synthetic cannabinoids are known to have an affinity for both CB1R and CB2R, they, like natural cannabinoids, can also interact with non-CBR, such as vanilloid or serotonergic receptors [89]. The most common group of synthetic cannabinoids is JWH, where JWH-018 has potent pharmacological activity and can be easily synthesized and used to synthesize other synthetic cannabinoids with different properties and affinities for CBR [90]. Besides JWH, other common groups of synthetic cannabinoids are HU and CP groups. While HU are classic cannabinoids, CP are cannabimimetics originally developed by Pfizer in the 1970s [90]. Some of the most extensively studied selective CB1 or mixed CB1/CB2 agonists are WIN 55,212-2, HU 210, ACEA, and JWH-018 [91]. In addition, several synthetic selective CB2R agonists, such as GSK554418A, GW833972A, GW842166X, HU-308, GW405833, JWH-015, JWH-133, A-836339, AM1241, AM630, NESS400, etc., have been reported in the literature and some (Cannabinir, GW842166, Tedalinab, GRC10693, S-7774698, LY2828360, KHK6188, Lenabasum) are under investigation at various stages of clinical development [85][89][85,89]. In humans, CB2R are encoded by the cannabinoid receptor 2 (CNR2) gene, which is located on chromosome 1p36 and consists of 360 amino acids [92]. The CB2R share 44% total amino acid homology and 68% homology in the transmembrane domains with the CB1R [92]. The CB2R were cloned in 1993, and these receptors were previously thought to be absent from the brain, as they were only detectable in the periphery [51][93][94][51,93,94]. In contrast to CB1R, which are mainly found in the CNS, particularly in presynaptic neurons at central and peripheral nerve terminals, where they inhibit neurotransmitter release [95], CB2R predominate in cells and tissues involved in the immune response, such as the spleen, thymus and blood-derived monocytes [51][96][51,96], and modulate interleukin release and cell migration. Until recently, the significant increase of CB2R in the CNS was thought to occur specifically in activated microglial cells under inflammatory conditions but was not measurable under physiological conditions or in other brain cell types [97]. However, using methods such as immunostaining, in situ hybridization, and gene expression analysis, CB2R has been shown to be present throughout different brain regions [98][99][100][101][98,99,100,101], including the striatum, amygdala, hippocampus, cortex and ventral tegmental area [102], in neural progenitor cells, neurons, as well as glial and endothelial cells [103][104][105][106][103,104,105,106]. In neurons, CB2Rs appear to be mainly distributed in postsynaptic somatodendritic regions, and their activation inhibits neuronal excitability through membrane hyperpolarization [97][98][101][97,98,101]. Novel detection techniques allowed more precise detection of low CB2R mRNA levels, specifically in astrocytes, dopaminergic, glutamatergic and GABAergic neurons, but not in resting microglia [11][107][108][109][11,107,108,109]. Although there are numerous studies on the regulation of CB1R, knowledge of the physiological and pathological role of CB2R is limited. Activation of CB2R leads to the inhibition of neuroinflammatory signaling pathways, as well as a return from the pro-inflammatory state of microglia to normal anti-inflammatory function [85]. Thus, in in vitro experiments, AEA has been shown to act via the MAPK signaling pathway within the CNS immune system to reduce the magnitude of the inflammatory response, as well as to limit neurodegenerative immune responses [110]. Moreover, AEA was found to reduce lipopolysaccharide-induced neuroinflammation in primary rat microglial cultures [111]. Even though AEA can activate CB1R, CB2R and other receptors of the ECS, the anti-inflammatory actions appear to be mediated by CB2R [112]. Therefore, AEA may have a potential therapeutic effect on microglial-derived neuroinflammation and regulate many aspects of the inflammatory response in the brain. However, since CB2R ligands exert neuroprotection without psychotropic effects (strong mood alterations, anxiety, acute psychosis, cognitive and motor impairments), usually seen with CB1R agonists [95], new and selective CB2R ligands may be promising and safe drugs for the treatment of various neuroinflammatory disorders [111]. Nevertheless, CB2R agonists also have disadvantages, such as immune suppression during chronic use, or pro-inflammatory actions [111]. Only a few synthetic CB2R agonists have reached clinical trials, despite increasing reports of selective CB2R ligands and high expectations with these ECS targets [113]. Some of them, such as GW842166X, CP55940, S-777469 and JTE-907, have already completed Phase II trials in various pain disorders; however, none of them have been assessed for neurodegenerative or neuroinflammatory disorders in humans [111]. Recently, new CB2R ligands have been characterized for their potential neuroprotective effects and the most prominent among them, the inverse agonist of CB2R SMM-189 seems to achieve neuroprotection by modulating microglial activation in a mouse model of mild traumatic brain injury [114]. Specifically, SMM-189 reduces some pro-inflammatory markers, indicating decreased infiltration of peripheral macrophages and other immune cells involved in neurodegeneration [115]. Recently, new strategies targeting CB2R for neurodegenerative and neuroinflammatory disorders have emerged when 4′-O-methylhokiol, the main bioactive component of Magnolia grandiflora L. that acts as both CB2R modulator and COX-2 substrate-specific inhibitor, has shown beneficial effects in animal models of neurodegeneration [116]. Moreover, targeting CB2R homo- and heterodimers needs to be further investigated [111]. While homobivalent and heterobivalent CB1R ligands have been previously designed and described in the literature [117], the first structurally bivalent CB2R compounds were designed and synthesized in 2014; however, with lower activity and selectivity compared to their monomeric counterparts [118]. Whereas monomeric compound is a selective CB2R agonists, bivalent compounds are weak antagonists/inverse agonists at CB1R and CB2R [118]. Another therapeutic possibility is the use of ligand-biased signaling [119]. For example, 2-AG is a very potent activator of the ERK1/2-MAPK signaling pathway at low concentrations, although higher concentrations are required to inhibit the adenylyl cyclase and calcium pathways [120]. In the future, CB2R allosteric modulators may offer new therapeutic approaches due to their potential to fine-tune receptor responses while minimizing the side effects [111]. Currently, the allosteric modulation specific to the CB2R signaling is still evaluated [121], while CB2R positive and negative allosteric modulators remain to be discovered.5. Role of CB2R in Various CNS Disorders
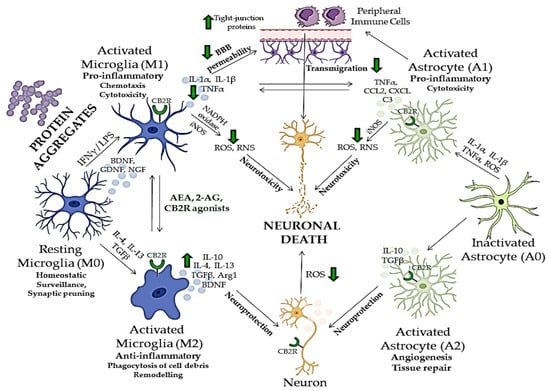
Neurodegenerative disorders, such as Alzheimer’s disease, Parkinson’s disease, Huntington’s disease, multiple sclerosis and amyotrophic lateral sclerosis, are characterized by a progressive loss of specific neurons in various brain regions, which leads to different symptomatic and clinical outcomes [122][123][122,123]. Since there is no cure for these diseases, therapeutic approaches mostly consist of partial symptomatic relief, which do not halt the progression of the disease. The main hallmarks of neurodegenerative diseases are neuroinflammation, oxidative stress, abnormal protein accumulation and excitotoxicity [124]. Studies have shown that pharmacological modulation of endocannabinoid signaling can modulate these neurodegeneration traits and cause alleviation of symptoms and disease progression [125]. Targeting different components of ECS, therefore, brings new aspects to understanding mechanisms underlying different CNS disorders to provide novel, more effective therapies. The role of CB1R in behavior, emotions, learning and memory, addiction and various other CNS disorders has been widely studied [49][126][49,126]. Previous studies have shown that dysregulation of CB1R in different CNS regions is involved in the pathophysiology of schizophrenia, MDD and anxiety disorders [127][128][129][127,128,129]. Therefore, normalizing the CB1R activity may have beneficial effects in treating these disorders. However, obtained findings demonstrated that CB1R pharmacological targeting induces serious side effects, such as depression, psychosis, panic attacks, anxiety and even suicidal ideation [130][131][130,131]. Hence, there is an emerging need to study new therapeutic targets with minimal adverse effects [130][131][130,131]. Recent studies suggested that targeting CB2R in CNS is effective and safe and may open a new possibility for the modulation of ECS. Compared to CB1R, CB2R have lower expression levels in the brain under normal physiological conditions, but their enhanced levels were observed in neurodegenerative and neuropsychiatric disorders [101][105][132][101,105,132]. Recent findings suggested that B2R modulates the behavioral effects in the CNS [9], including mood and emotional behavior. Evidence suggests CB2R plays a role in food intake, body weight control and eating disorders [133][134][135][133,134,135], depression and anxiety [101][105][134][101,105,134], drug addiction [136], psychosis and schizophrenia-like behavior [137][138][139][137,138,139] and synaptic plasticity underlying cognitive functions [135][138][135,138]. Elevated CB2R expression levels have been reported in several pathological conditions, such as neurological pain [136][140][136,140], stroke [137], traumatic brain injury [140][141][140,141], addiction [142][143][142,143] and neurodegenerative diseases, including multiple sclerosis [144][145][144,145]. CB2R anti-inflammatory action has been found in animal studies and in experiments using cell cultures [141][146][141,146]. The activation of CB2R decreases neuroinflammation, partly by mediating the transition of microglial phenotype from a predominantly neurotoxic ‘’M1” to a neuroprotective ‘’M2” [147], suggesting an important role of CB2R in restoring homeostasis [85]. Therefore, due to CBR2 inducible nature during inflammation, ligands that activate or inhibit their activity could be used for potential therapeutic purposes in various CNS disorders whose pathogenesis involves neuroinflammatory processes [97].6. Role of CB2R in Neuroinflammation and Neurodegeneration
A strong relationship between neuroinflammation and neurodegeneration has been reported in the early stages of neurodegenerative disorders, such as Alzheimer’s disease, frontotemporal dementia, Parkinson’s disease, amyotrophic lateral sclerosis and Huntington’s disease. This link is also strong in primarily inflammatory diseases such as multiple sclerosis and human immunodeficiency virus (HIV)-associated dementia associated with intense and chronic inflammation of myelin sheets and HIV infection of microglia, respectively, consequently leading to neuronal damage [123]. Moreover, the neuroinflammatory condition is characteristic of other psychiatric disorders and neurological diseases such as epilepsy, and traumatic brain injury, where it mediates secondary neurodegeneration [148][149][148,149]. Many aspects of neuroinflammation and neurodegeneration cross-talk remain unclear; however, recent studies showed that glial cells, especially microglia, which act as the brain’s immune cells, could be crucial mediators of neurodegeneration, together with peripheral monocytes which cross BBB under CNS pathological conditions (Figure 2) [150][151][152][150,151,152]. ECS has been, therefore, extensively investigated in relation to neurodegenerative and neuroinflammatory mechanisms of CNS disorders, and potentially novel treatment strategies [153]. Although there are major differences in the etiology, physiology and clinical picture of various neurodegenerative proteinopathies, aggregation and accumulation of defected and misfolded proteins such as Aβ, hyperphosphorylated tau [154], α-synuclein [155], mutated superoxidase dismutase 1 (mSOD1) [156] and huntingtin [157], are shared aspects of these disorders and all represent activation stimulus to circulating microglia [158].