 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Francesco Boccalatte | -- | 3499 | 2022-12-06 22:13:47 | | | |
| 2 | Lindsay Dong | + 3 word(s) | 3502 | 2022-12-08 01:53:22 | | | | |
| 3 | Lindsay Dong | -1 word(s) | 3501 | 2022-12-08 08:41:58 | | |
Video Upload Options
Chimeric antigen receptor (CAR) T cells are genetically engineered T cells that recognize markers present on tumor cells and drive the degradation of the tumor itself. CAR T immunotherapy has obtained remarkable success in targeting a number of blood malignancies; however, its outcome is typically modest when applied to solid tumors, because of specific structural, biological, and metabolic aspects of the solid tumor environment.
1. The Solid Tumor Microenvironment and Its Impact on CAR T Therapy
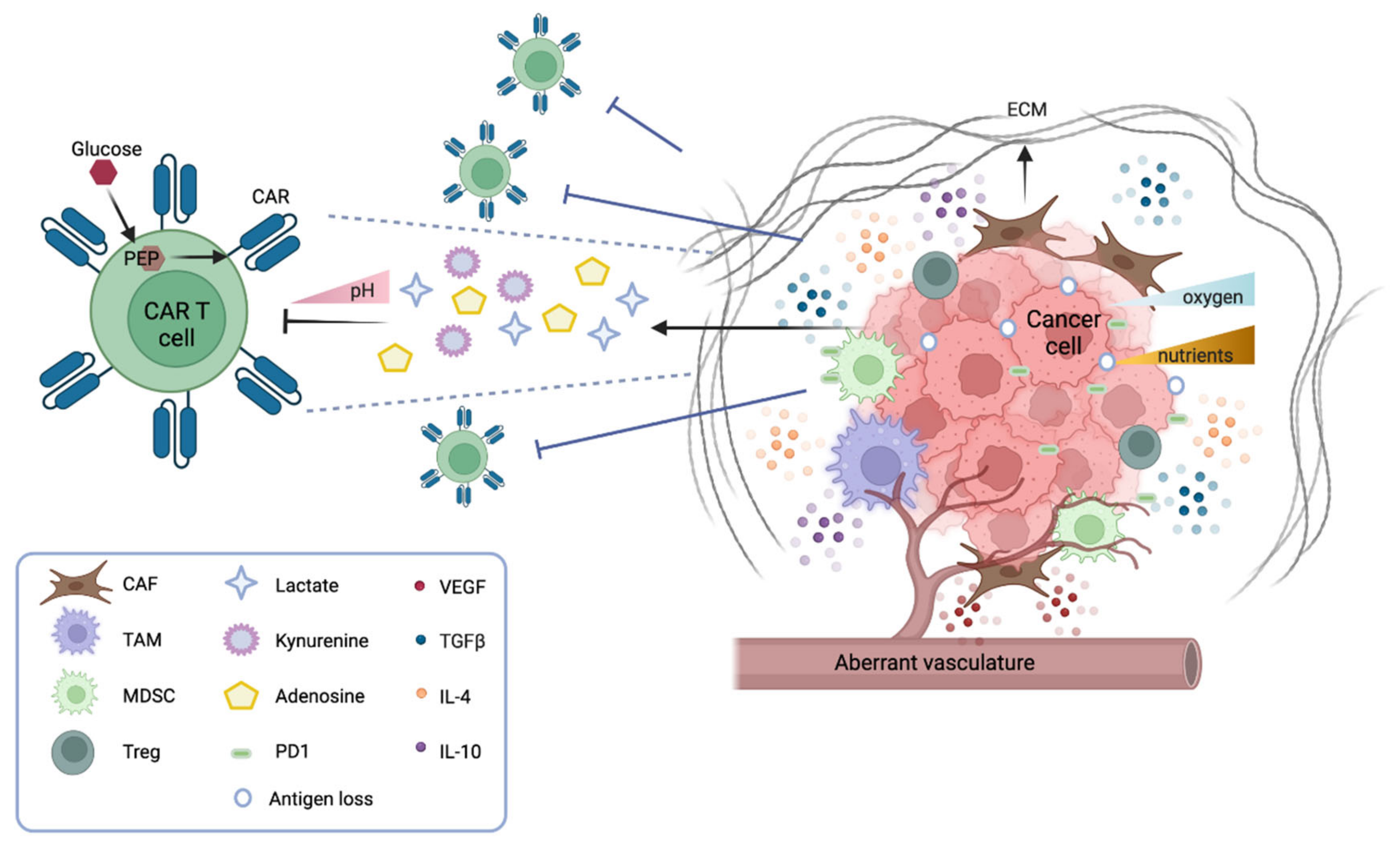
1.1. CAR T Cell Trafficking and Infiltration
1.2. The Solid Tumor Microenvironment: Physical and Metabolic Barriers
Several physical barriers hamper the accessibility of CAR T cells to a solid tumor mass, including thick surrounding tumor stroma, aberrant vasculature, and high interstitial pressure. The stroma is mostly constituted by cancer-associated fibroblasts (CAFs) that drive the deposition of the extracellular matrix (ECM), thus physically preventing the infiltration of immune cells. Several strategies are under investigation to overcome this obstacle. CAR T cells can be engineered to target fibroblast activation protein (FAP), therefore reducing the number of CAFs in the microenvironment [2]. Alternatively, CAR T cells can be armored with proteases that degrade the ECM. For instance, CAR T cells engineered to express heparanase, which degrades the heparan sulfate component of the ECM, have shown better infiltration and tumor clearance both in vitro and in animal models [3]. Moreover, the aberrant tumor vasculature causes interstitial hypertension that prevents extravasation and a hypoxic microenvironment, especially in the central part of the tumor. Thus, normalizing the tumor vasculature may be beneficial [4]. In this context, vascular endothelial growth factor (VEGF) signaling plays a pivotal role. Antiangiogenic therapy that blocks VEGF signaling improves immune cell infiltration [5], and anti-VEGFR CAR T cells can efficiently inhibit tumor growth—as shown in several syngeneic mouse models [6].
1.3. The Solid Tumor Microenvironment: Soluble and Cellular Drivers of Immune Suppression
2. CAR T Cell Immune Therapy for Solid Tumors
2.1. Toxicities
2.2. Gastrointestinal Cancers
2.3. Genitourinary Cancers and Beyond
2.4. Brain Tumors
2.5. Pediatric Sarcomas
3. Conclusions
References
- Beatty, G.L.; Gladney, W.L. Immune escape mechanisms as a guide for cancer immunotherapy. Clin. Cancer Res. 2015, 21, 687–692.
- Wang, L.C.; Lo, A.; Scholler, J.; Sun, J.; Majumdar, R.S.; Kapoor, V.; Antzis, M.; Cotner, C.E.; Johnson, L.A.; Durham, A.C.; et al. Targeting fibroblast activation protein in tumor stroma with chimeric antigen receptor T cells can inhibit tumor growth and augment host immunity without severe toxicity. Cancer Immunol. Res. 2014, 2, 154–166.
- Caruana, I.; Savoldo, B.; Hoyos, V.; Weber, G.; Liu, H.; Kim, E.S.; Ittmann, M.M.; Marchetti, D.; Dotti, G. Heparanase promotes tumor infiltration and antitumor activity of CAR-redirected T lymphocytes. Nat. Med. 2015, 21, 524–529.
- Newport, E.L.; Pedrosa, A.R.; Njegic, A.; Hodivala-Dilke, K.M.; Munoz-Felix, J.M. Improved Immunotherapy Efficacy by Vascular Modulation. Cancers 2021, 13, 5207.
- Allen, E.; Jabouille, A.; Rivera, L.B.; Lodewijckx, I.; Missiaen, R.; Steri, V.; Feyen, K.; Tawney, J.; Hanahan, D.; Michael, I.P.; et al. Combined antiangiogenic and anti-PD-L1 therapy stimulates tumor immunity through HEV formation. Sci. Transl. Med. 2017, 9, 385.
- Chinnasamy, D.; Yu, Z.; Theoret, M.R.; Zhao, Y.; Shrimali, R.K.; Morgan, R.A.; Feldman, S.A.; Restifo, N.P.; Rosenberg, S.A. Gene therapy using genetically modified lymphocytes targeting VEGFR-2 inhibits the growth of vascularized syngenic tumors in mice. J. Clin. Investig. 2010, 120, 3953–3968.
- Berahovich, R.; Liu, X.; Zhou, H.; Tsadik, E.; Xu, S.; Golubovskaya, V.; Wu, L. Hypoxia Selectively Impairs CAR-T Cells In Vitro. Cancers 2019, 11, 602.
- Juillerat, A.; Marechal, A.; Filhol, J.M.; Valogne, Y.; Valton, J.; Duclert, A.; Duchateau, P.; Poirot, L. An oxygen sensitive self-decision making engineered CAR T-cell. Sci. Rep. 2017, 7, 39833.
- Chang, C.H.; Curtis, J.D.; Maggi, L.B.; Faubert, B., Jr.; Villarino, A.V.; O’Sullivan, D.; Huang, S.C.; van der Windt, G.J.; Blagih, J.; Qiu, J.; et al. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell 2013, 153, 1239–1251.
- Ho, P.C.; Bihuniak, J.D.; Macintyre, A.N.; Staron, M.; Liu, X.; Amezquita, R.; Tsui, Y.C.; Cui, G.; Micevic, G.; Perales, J.C.; et al. Phosphoenolpyruvate Is a Metabolic Checkpoint of Anti-tumor T Cell Responses. Cell 2015, 162, 1217–1228.
- Kawalekar, O.U.; O’Connor, R.S.; Fraietta, J.A.; Guo, L.; McGettigan, S.E.; Posey, A.D., Jr.; Patel, P.R.; Guedan, S.; Scholler, J.; Keith, B.; et al. Distinct Signaling of Coreceptors Regulates Specific Metabolism Pathways and Impacts Memory Development in CAR T Cells. Immunity 2016, 44, 712.
- Beavis, P.A.; Henderson, M.A.; Giuffrida, L.; Mills, J.K.; Sek, K.; Cross, R.S.; Davenport, A.J.; John, L.B.; Mardiana, S.; Slaney, C.Y.; et al. Targeting the adenosine 2A receptor enhances chimeric antigen receptor T cell efficacy. J. Clin. Investig. 2017, 127, 929–941.
- Goodwin, J.S.; Bankhurst, A.D.; Messner, R.P. Suppression of human T-cell mitogenesis by prostaglandin. Existence of a prostaglandin-producing suppressor cell. J. Exp. Med. 1977, 146, 1719–1734.
- Newick, K.; O’Brien, S.; Sun, J.; Kapoor, V.; Maceyko, S.; Lo, A.; Pure, E.; Moon, E.; Albelda, S.M. Augmentation of CAR T-cell Trafficking and Antitumor Efficacy by Blocking Protein Kinase A Localization. Cancer Immunol. Res. 2016, 4, 541–551.
- Chakravarthy, A.; Khan, L.; Bensler, N.P.; Bose, P.; De Carvalho, D.D. TGF-beta-associated extracellular matrix genes link cancer-associated fibroblasts to immune evasion and immunotherapy failure. Nat. Commun. 2018, 9, 4692.
- Oh, S.A.; Li, M.O. TGF-beta: Guardian of T cell function. J. Immunol. 2013, 191, 3973–3979.
- Wallace, A.; Kapoor, V.; Sun, J.; Mrass, P.; Weninger, W.; Heitjan, D.F.; June, C.; Kaiser, L.R.; Ling, L.E.; Albelda, S.M. Transforming growth factor-beta receptor blockade augments the effectiveness of adoptive T-cell therapy of established solid cancers. Clin. Cancer Res. 2008, 14, 3966–3974.
- Bollard, C.M.; Rossig, C.; Calonge, M.J.; Huls, M.H.; Wagner, H.J.; Massague, J.; Brenner, M.K.; Heslop, H.E.; Rooney, C.M. Adapting a transforming growth factor beta-related tumor protection strategy to enhance antitumor immunity. Blood 2002, 99, 3179–3187.
- Hou, A.J.; Chang, Z.L.; Lorenzini, M.H.; Zah, E.; Chen, Y.Y. TGF-beta-responsive CAR-T cells promote anti-tumor immune function. Bioeng. Transl. Med. 2018, 3, 75–86.
- Wilkie, S.; Burbridge, S.E.; Chiapero-Stanke, L.; Pereira, A.C.; Cleary, S.; van der Stegen, S.J.; Spicer, J.F.; Davies, D.M.; Maher, J. Selective expansion of chimeric antigen receptor-targeted T-cells with potent effector function using interleukin-4. J. Biol. Chem. 2010, 285, 25538–25544.
- Leen, A.M.; Sukumaran, S.; Watanabe, N.; Mohammed, S.; Keirnan, J.; Yanagisawa, R.; Anurathapan, U.; Rendon, D.; Heslop, H.E.; Rooney, C.M.; et al. Reversal of tumor immune inhibition using a chimeric cytokine receptor. Mol. Ther. 2014, 22, 1211–1220.
- Sukumaran, S.; Watanabe, N.; Bajgain, P.; Raja, K.; Mohammed, S.; Fisher, W.E.; Brenner, M.K.; Leen, A.M.; Vera, J.F. Enhancing the Potency and Specificity of Engineered T Cells for Cancer Treatment. Cancer Discov. 2018, 8, 972–987.
- Chmielewski, M.; Hombach, A.A.; Abken, H. Of CARs and TRUCKs: Chimeric antigen receptor (CAR) T cells engineered with an inducible cytokine to modulate the tumor stroma. Immunol. Rev. 2014, 257, 83–90.
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437.
- Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Bronte, V. Coordinated regulation of myeloid cells by tumours. Nat. Rev. Immunol. 2012, 12, 253–268.
- Hamanishi, J.; Mandai, M.; Matsumura, N.; Abiko, K.; Baba, T.; Konishi, I. PD-1/PD-L1 blockade in cancer treatment: Perspectives and issues. Int. J. Clin. Oncol. 2016, 21, 462–473.
- Cherkassky, L.; Morello, A.; Villena-Vargas, J.; Feng, Y.; Dimitrov, D.S.; Jones, D.R.; Sadelain, M.; Adusumilli, P.S. Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J. Clin. Investig. 2016, 126, 3130–3144.
- Sojka, D.K.; Huang, Y.H.; Fowell, D.J. Mechanisms of regulatory T-cell suppression—A diverse arsenal for a moving target. Immunology 2008, 124, 13–22.
- Kim, J.H.; Kim, B.S.; Lee, S.K. Regulatory T Cells in Tumor Microenvironment and Approach for Anticancer Immunotherapy. Immune. Netw. 2020, 20, e4.
- Lee, D.W.; Santomasso, B.D.; Locke, F.L.; Ghobadi, A.; Turtle, C.J.; Brudno, J.N.; Maus, M.V.; Park, J.H.; Mead, E.; Pavletic, S.; et al. ASTCT Consensus Grading for Cytokine Release Syndrome and Neurologic Toxicity Associated with Immune Effector Cells. Biol. Blood Marrow. Transpl. 2019, 25, 625–638.
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, E359–E386.
- Zhang, J.; Dong, R.; Shen, L. Evaluation and reflection on claudin 18.2 targeting therapy in advanced gastric cancer. Chin. J. Cancer Res. 2020, 32, 263–270.
- Cho, H.S.; Mason, K.; Ramyar, K.X.; Stanley, A.M.; Gabelli, S.B.; Denney, D.W., Jr.; Leahy, D.J. Structure of the extracellular region of HER2 alone and in complex with the Herceptin Fab. Nature 2003, 421, 756–760.
- Boku, N. HER2-positive gastric cancer. Gastric. Cancer 2014, 17, 1–12.
- Holzinger, A.; Abken, H. CAR T cells targeting solid tumors: Carcinoembryonic antigen (CEA) proves to be a safe target. Cancer Immunol. Immunother. 2017, 66, 1505–1507.
- Beatty, G.L.; O’Hara, M. Chimeric antigen receptor-modified T cells for the treatment of solid tumors: Defining the challenges and next steps. Pharmacol. Ther. 2016, 166, 30–39.
- Beatty, G.L.; O’Hara, M.H.; Lacey, S.F.; Torigian, D.A.; Nazimuddin, F.; Chen, F.; Kulikovskaya, I.M.; Soulen, M.C.; McGarvey, M.; Nelson, A.M.; et al. Activity of Mesothelin-Specific Chimeric Antigen Receptor T Cells Against Pancreatic Carcinoma Metastases in a Phase 1 Trial. Gastroenterology 2018, 155, 29–32.
- Adusumilli, P.S.; Zauderer, M.G.; Riviere, I.; Solomon, S.B.; Rusch, V.W.; O’Cearbhaill, R.E.; Zhu, A.; Cheema, W.; Chintala, N.K.; Halton, E.; et al. A Phase I Trial of Regional Mesothelin-Targeted CAR T-cell Therapy in Patients with Malignant Pleural Disease, in Combination with the Anti-PD-1 Agent Pembrolizumab. Cancer Discov. 2021, 11, 2748–2763.
- The Lancet Oncology. CAR T-cell therapy for solid tumours. Lancet Oncol. 2021, 22, 893.
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996.
- Soler, D.C.; Kerstetter-Fogle, A.; McCormick, T.S.; Sloan, A.E. Using chimeric antigen receptor T-cell therapy to fight glioblastoma multiforme: Past, present and future developments. J. Neurooncol. 2022, 156, 81–96.
- Brown, C.E.; Alizadeh, D.; Starr, R.; Weng, L.; Wagner, J.R.; Naranjo, A.; Ostberg, J.R.; Blanchard, M.S.; Kilpatrick, J.; Simpson, J.; et al. Regression of Glioblastoma after Chimeric Antigen Receptor T-Cell Therapy. N. Engl. J. Med. 2016, 375, 2561–2569.
- Majzner, R.G.; Ramakrishna, S.; Yeom, K.W.; Patel, S.; Chinnasamy, H.; Schultz, L.M.; Richards, R.M.; Jiang, L.; Barsan, V.; Mancusi, R.; et al. GD2-CAR T cell therapy for H3K27M-mutated diffuse midline gliomas. Nature 2022, 603, 934–941.
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110.
- Brennan, C.W.; Verhaak, R.G.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462–477.
- Neftel, C.; Laffy, J.; Filbin, M.G.; Hara, T.; Shore, M.E.; Rahme, G.J.; Richman, A.R.; Silverbush, D.; Shaw, M.L.; Hebert, C.M.; et al. An Integrative Model of Cellular States, Plasticity, and Genetics for Glioblastoma. Cell 2019, 178, 835–849.
- Patel, A.P.; Tirosh, I.; Trombetta, J.J.; Shalek, A.K.; Gillespie, S.M.; Wakimoto, H.; Cahill, D.P.; Nahed, B.V.; Curry, W.T.; Martuza, R.L.; et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 2014, 344, 1396–1401.
- Snuderl, M.; Fazlollahi, L.; Le, L.P.; Nitta, M.; Zhelyazkova, B.H.; Davidson, C.J.; Akhavanfard, S.; Cahill, D.P.; Aldape, K.D.; Betensky, R.A.; et al. Mosaic amplification of multiple receptor tyrosine kinase genes in glioblastoma. Cancer Cell 2011, 20, 810–817.
- Thokala, R.; Binder, Z.A.; Yin, Y.; Zhang, L.; Zhang, J.V.; Zhang, D.Y.; Milone, M.C.; Ming, G.L.; Song, H.; O’Rourke, D.M. High-Affinity Chimeric Antigen Receptor With Cross-Reactive scFv to Clinically Relevant EGFR Oncogenic Isoforms. Front. Oncol. 2021, 11, 664236.
- Heimberger, A.B.; Hlatky, R.; Suki, D.; Yang, D.; Weinberg, J.; Gilbert, M.; Sawaya, R.; Aldape, K. Prognostic effect of epidermal growth factor receptor and EGFRvIII in glioblastoma multiforme patients. Clin. Cancer Res. 2005, 11, 1462–1466.
- Sampson, J.H.; Choi, B.D.; Sanchez-Perez, L.; Suryadevara, C.M.; Snyder, D.J.; Flores, C.T.; Schmittling, R.J.; Nair, S.K.; Reap, E.A.; Norberg, P.K.; et al. EGFRvIII mCAR-modified T-cell therapy cures mice with established intracerebral glioma and generates host immunity against tumor-antigen loss. Clin. Cancer Res. 2014, 20, 972–984.
- Lowenstein, P.R.; Castro, M.G. The value of EGFRvIII as the target for glioma vaccines. Am. Soc. Clin. Oncol. Educ. Book 2014, 34, 42–50.
- Rosenthal, M.; Curry, R.; Reardon, D.A.; Rasmussen, E.; Upreti, V.V.; Damore, M.A.; Henary, H.A.; Hill, J.S.; Cloughesy, T. Safety, tolerability, and pharmacokinetics of anti-EGFRvIII antibody-drug conjugate AMG 595 in patients with recurrent malignant glioma expressing EGFRvIII. Cancer Chemother. Pharmacol. 2019, 84, 327–336.
- Sampson, J.H.; Archer, G.E.; Mitchell, D.A.; Heimberger, A.B.; Bigner, D.D. Tumor-specific immunotherapy targeting the EGFRvIII mutation in patients with malignant glioma. Semin. Immunol. 2008, 20, 267–275.
- Cui, X.; Ma, C.; Vasudevaraja, V.; Serrano, J.; Tong, J.; Peng, Y.; Delorenzo, M.; Shen, G.; Frenster, J.; Morales, R.T.; et al. Dissecting the immunosuppressive tumor microenvironments in Glioblastoma-on-a-Chip for optimized PD-1 immunotherapy. eLife 2020, 9, e52253.
- Grabowski, M.M.; Sankey, E.W.; Ryan, K.J.; Chongsathidkiet, P.; Lorrey, S.J.; Wilkinson, D.S.; Fecci, P.E. Immune suppression in gliomas. J. Neurooncol. 2021, 151, 3–12.
- Woroniecka, K.I.; Rhodin, K.E.; Chongsathidkiet, P.; Keith, K.A.; Fecci, P.E. T-cell Dysfunction in Glioblastoma: Applying a New Framework. Clin. Cancer Res. 2018, 24, 3792–3802.
- Bayin, N.S.; Ma, L.; Thomas, C.; Baitalmal, R.; Sure, A.; Fansiwala, K.; Bustoros, M.; Golfinos, J.G.; Pacione, D.; Snuderl, M.; et al. Patient-Specific Screening Using High-Grade Glioma Explants to Determine Potential Radiosensitization by a TGF-beta Small Molecule Inhibitor. Neoplasia 2016, 18, 795–805.
- Barcellos-Hoff, M.H.; Newcomb, E.W.; Zagzag, D.; Narayana, A. Therapeutic targets in malignant glioblastoma microenvironment. Semin. Radiat. Oncol. 2009, 19, 163–170.
- Hardee, M.E.; Marciscano, A.E.; Medina-Ramirez, C.M.; Zagzag, D.; Narayana, A.; Lonning, S.M.; Barcellos-Hoff, M.H. Resistance of glioblastoma-initiating cells to radiation mediated by the tumor microenvironment can be abolished by inhibiting transforming growth factor-beta. Cancer Res. 2012, 72, 4119–4129.
- Magana-Maldonado, R.; Chavez-Cortez, E.G.; Olascoaga-Arellano, N.K.; Lopez-Mejia, M.; Maldonado-Leal, F.M.; Sotelo, J.; Pineda, B. Immunological Evasion in Glioblastoma. BioMed Res. Int. 2016, 2016, 7487313.
- Crane, C.A.; Han, S.J.; Barry, J.J.; Ahn, B.J.; Lanier, L.L.; Parsa, A.T. TGF-beta downregulates the activating receptor NKG2D on NK cells and CD8+ T cells in glioma patients. Neuro. Oncol. 2010, 12, 7–13.
- Leitlein, J.; Aulwurm, S.; Waltereit, R.; Naumann, U.; Wagenknecht, B.; Garten, W.; Weller, M.; Platten, M. Processing of immunosuppressive pro-TGF-beta 1,2 by human glioblastoma cells involves cytoplasmic and secreted furin-like proteases. J. Immunol. 2001, 166, 7238–7243.
- Bodmer, S.; Strommer, K.; Frei, K.; Siepl, C.; de Tribolet, N.; Heid, I.; Fontana, A. Immunosuppression and transforming growth factor-beta in glioblastoma. Preferential production of transforming growth factor-beta 2. J. Immunol. 1989, 143, 3222–3229.
- Cui, X.; Morales, R.T.; Qian, W.; Wang, H.; Gagner, J.P.; Dolgalev, I.; Placantonakis, D.; Zagzag, D.; Cimmino, L.; Snuderl, M.; et al. Hacking macrophage-associated immunosuppression for regulating glioblastoma angiogenesis. Biomaterials 2018, 161, 164–178.
- Close, H.J.; Stead, L.F.; Nsengimana, J.; Reilly, K.A.; Droop, A.; Wurdak, H.; Mathew, R.K.; Corns, R.; Newton-Bishop, J.; Melcher, A.A.; et al. Expression profiling of single cells and patient cohorts identifies multiple immunosuppressive pathways and an altered NK cell phenotype in glioblastoma. Clin. Exp. Immunol. 2020, 200, 33–44.
- Chongsathidkiet, P.; Jackson, C.; Koyama, S.; Loebel, F.; Cui, X.; Farber, S.H.; Woroniecka, K.; Elsamadicy, A.A.; Dechant, C.A.; Kemeny, H.R.; et al. Sequestration of T cells in bone marrow in the setting of glioblastoma and other intracranial tumors. Nat. Med. 2018, 24, 1459–1468.
- Suryadevara, C.M.; Desai, R.; Farber, S.H.; Choi, B.D.; Swartz, A.M.; Shen, S.H.; Gedeon, P.C.; Snyder, D.J.; Herndon, J.E., 2nd; Healy, P.; et al. Preventing Lck Activation in CAR T Cells Confers Treg Resistance but Requires 4-1BB Signaling for Them to Persist and Treat Solid Tumors in Nonlymphodepleted Hosts. Clin. Cancer Res. 2019, 25, 358–368.
- Kakarla, S.; Chow, K.K.; Mata, M.; Shaffer, D.R.; Song, X.T.; Wu, M.F.; Liu, H.; Wang, L.L.; Rowley, D.R.; Pfizenmaier, K.; et al. Antitumor effects of chimeric receptor engineered human T cells directed to tumor stroma. Mol. Ther. 2013, 21, 1611–1620.
- Rodriguez-Garcia, A.; Lynn, R.C.; Poussin, M.; Eiva, M.A.; Shaw, L.C.; O’Connor, R.S.; Minutolo, N.G.; Casado-Medrano, V.; Lopez, G.; Matsuyama, T.; et al. CAR-T cell-mediated depletion of immunosuppressive tumor-associated macrophages promotes endogenous antitumor immunity and augments adoptive immunotherapy. Nat. Commun. 2021, 12, 877.
- Da Mesquita, S.; Louveau, A.; Vaccari, A.; Smirnov, I.; Cornelison, R.C.; Kingsmore, K.M.; Contarino, C.; Onengut-Gumuscu, S.; Farber, E.; Raper, D.; et al. Functional aspects of meningeal lymphatics in ageing and Alzheimer’s disease. Nature 2018, 560, 185–191.
- Ahn, J.H.; Cho, H.; Kim, J.H.; Kim, S.H.; Ham, J.S.; Park, I.; Suh, S.H.; Hong, S.P.; Song, J.H.; Hong, Y.K.; et al. Meningeal lymphatic vessels at the skull base drain cerebrospinal fluid. Nature 2019, 572, 62–66.
- Song, E.; Mao, T.; Dong, H.; Boisserand, L.S.B.; Antila, S.; Bosenberg, M.; Alitalo, K.; Thomas, J.L.; Iwasaki, A. VEGF-C-driven lymphatic drainage enables immunosurveillance of brain tumours. Nature 2020, 577, 689–694.
- Da Mesquita, S.; Papadopoulos, Z.; Dykstra, T.; Brase, L.; Farias, F.G.; Wall, M.; Jiang, H.; Kodira, C.D.; de Lima, K.A.; Herz, J.; et al. Meningeal lymphatics affect microglia responses and anti-Abeta immunotherapy. Nature 2021, 593, 255–260.
- Jafari, F.; Javdansirat, S.; Sanaie, S.; Naseri, A.; Shamekh, A.; Rostamzadeh, D.; Dolati, S. Osteosarcoma: A comprehensive review of management and treatment strategies. Ann. Diagn. Pathol. 2020, 49, 151654.
- Suri, M.; Soni, N.; Okpaleke, N.; Yadav, S.; Shah, S.; Iqbal, Z.; Alharbi, M.G.; Kalra, H.S.; Hamid, P. A Deep Dive Into the Newest Avenues of Immunotherapy for Pediatric Osteosarcoma: A Systematic Review. Cureus 2021, 13, e18349.
- Somers, G.R.; Ho, M.; Zielenska, M.; Squire, J.A.; Thorner, P.S. HER2 amplification and overexpression is not present in pediatric osteosarcoma: A tissue microarray study. Pediatr. Dev. Pathol. 2005, 8, 525–532.
- Li, Y.G.; Geng, X. A meta-analysis on the association of HER-2 overexpression with prognosis in human osteosarcoma. Eur. J. Cancer Care 2010, 19, 313–316.
- Lee, W.I.; Bacchni, P.; Bertoni, F.; Maeng, Y.H.; Park, Y.K. Quantitative assessment of HER2/neu expression by real-time PCR and fluorescent in situ hybridization analysis in low-grade osteosarcoma. Oncol. Rep. 2004, 12, 125–128.
- Gill, J.; Hingorani, P.; Roth, M.; Gorlick, R. HER2-Targeted Therapy in Osteosarcoma. Adv. Exp. Med. Biol. 2020, 1257, 55–66.
- Suzuki, M.; Cheung, N.K. Disialoganglioside GD2 as a therapeutic target for human diseases. Expert. Opin. Ther. Targets 2015, 19, 349–362.
- Chulanetra, M.; Morchang, A.; Sayour, E.; Eldjerou, L.; Milner, R.; Lagmay, J.; Cascio, M.; Stover, B.; Slayton, W.; Chaicumpa, W.; et al. GD2 chimeric antigen receptor modified T cells in synergy with sub-toxic level of doxorubicin targeting osteosarcomas. Am. J. Cancer Res. 2020, 10, 674–687.
- Majzner, R.G.; Theruvath, J.L.; Nellan, A.; Heitzeneder, S.; Cui, Y.; Mount, C.W.; Rietberg, S.P.; Linde, M.H.; Xu, P.; Rota, C.; et al. CAR T Cells Targeting B7-H3, a Pan-Cancer Antigen, Demonstrate Potent Preclinical Activity Against Pediatric Solid Tumors and Brain Tumors. Clin. Cancer Res. 2019, 25, 2560–2574.


