In the setting of solid tumors, immune cells should efficiently reach tumor sites and infiltrate masses that can be of considerable size. Successful immune cell trafficking depends on the concordant expression of chemokines secreted by the tumor, and the appropriate chemokine receptors on the T cells. Similarly, the infiltration process is driven by a matched expression of adhesion receptors/ligands by the T cells and the tumor endothelium. Unfortunately, tumors often downmodulate the expression of chemoattractant molecules, therefore escaping immune surveillance [
1].
1.2. The Solid Tumor Microenvironment: Physical and Metabolic Barriers
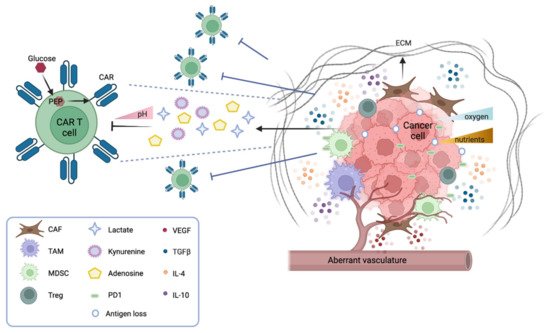
Several physical barriers hamper the accessibility of CAR T cells to a solid tumor mass, including thick surrounding tumor stroma, aberrant vasculature, and high interstitial pressure. The stroma is mostly constituted by cancer-associated fibroblasts (CAFs) that drive the deposition of the extracellular matrix (ECM), thus physically preventing the infiltration of immune cells. Several strategies are under investigation to overcome this obstacle. CAR T cells can be engineered to target fibroblast activation protein (FAP), therefore reducing the number of CAFs in the microenvironment [10]. Alternatively, CAR T cells can be armored with proteases that degrade the ECM. For instance, CAR T cells engineered to express heparanase, which degrades the heparan sulfate component of the ECM, have shown better infiltration and tumor clearance both in vitro and in animal models [11]. Moreover, the aberrant tumor vasculature causes interstitial hypertension that prevents extravasation and a hypoxic microenvironment, especially in the central part of the tumor. Thus, normalizing the tumor vasculature may be beneficial [12]. In this context, vascular endothelial growth factor (VEGF) signaling plays a pivotal role. Antiangiogenic therapy that blocks VEGF signaling improves immune cell infiltration [13], and anti-VEGFR CAR T cells can efficiently inhibit tumor growth—as shown in several syngeneic mouse models [14].
In terms of metabolic barriers, it is worth noticing that the particular anatomical structure of solid tumors generates hostile hypoxia and nutrient starvation for immune cells. The hypoxic environment caused by poor perfusion and abnormal vasculature hampers the expansion of CAR T cells and shifts their phenotype from effector to central memory [
15]. Strategies to favor CAR T response in the hypoxic TME are under investigation by fusing an oxygen-sensitive domain of hypoxia-inducible factor 1 (HIF1a) to the CAR scaffold [
16].
Of note, to mount an effective antitumor response, CAR T cells need to proliferate and produce cytokines and molecules that degrade tumor cells. Thus, CAR T cells must compete for nutrients and metabolites in a niche where tumor cells are scavenging most resources. CAR T cell effector functions rely on glucose and glycolytic metabolism [
17], which becomes highly challenging in a nutrient-poor environment. In particular, it has been observed that the insufficient production of phosphoenolpyruvate (PEP) in T cells can dampen TCR signaling, and therefore limit the effector response, and that PEP supplementation can efficiently restore T cell responses [
18]. It should also be noted that the addition of specific costimulatory molecules to the CAR structure has an impact on their glycolytic or fatty acid metabolism, and therefore their effectiveness in combating tumor cells [
19].
1.3. The Solid Tumor Microenvironment: Soluble and Cellular Drivers of Immune Suppression
The TME is replete with soluble factors released by both tumor and immune cells. Most of these factors have a direct suppressive role on CAR T cell adoptive immunotherapy. For example, adenosine is an immunosuppressive metabolite secreted by tumor and immune cells in the TME. In melanoma models, antagonists of the adenosine 2a receptor strongly increase the efficacy of CAR T therapy, either alone or in combination with a PD-1 checkpoint blockade [
24]. Another inhibitory factor is prostaglandin E2 (PGE2), an inflammatory molecule generated by tumor cells and macrophages that impairs CD4+ T cell proliferation and CD8+ T cell differentiation [
25]. Both PGE2 and adenosine exert their immunosuppressive function by activating protein kinase A (PKA), which inhibits TCR signaling. Newick et al. engineered CAR T cells to express a PKA inhibitor peptide, and showed that these armored CAR T cells had improved TCR signaling, cytokine production, and enhanced tumor killing [
26].
An important class of soluble factors in the TME is represented by cytokines and chemokines. These molecules can function either as boosters or inhibitors of antitumor responses. In the solid TME, cytokines act not only by impairing cytotoxic T cells, but also by recruiting immunosuppressor cells from peripheral sites, and by polarizing the resident immune cells towards an immunosuppressive phenotype. The most widely studied inhibitory cytokine in the context of the TME is tumor growth factor beta (TGFb). This factor acts both on the tumor stroma, where it enhances matrix deposition and shields tumor cells from immune surveillance [
27], and on T cells, where it inhibits effector functions and skews their phenotype towards immune tolerance [
28]. The systemic blockade of TGFb receptor signaling has been shown to enhance the efficacy of adoptive immunotherapy [
29]. Other studies have generated synthetic receptors to target TGFb signaling, such as a TGFb dominant negative (DN) receptor and a TGFb CAR. The TGFb DN receptor is a truncated, non-functional form of TGFb receptor that cannot transduce the intracellular signal, and therefore competes with the natural TGFb ligand–receptor function [
30]. The TGFb CAR has a double function, since it outcompetes the natural TGFb receptor for binding its ligand, and additionally stimulates the antitumor activity of neighboring cytotoxic T cells [
31].
Other inhibitory cytokines belong to the family of interleukins, for example IL-10 and IL-4. To counteract the inhibitory effect of IL-4, two different groups have engineered chimeric IL-4 receptors by fusing its extracellular domain with either the intracellular domain of the IL-2 receptor [
32] or the intracellular domain of the IL-7 receptor [
33]. These chimeric receptors can be combined with other approaches, and have been efficiently used to boost adoptive immunotherapy in animal models [
34]. Further studies have tried to increase the release of inflammatory cytokines, such as IL-12, in the TME to favor adoptive immunotherapy. CAR T cells that release IL-12 upon their activation can boost the natural immune cell response towards tumor cells that are escaping immunotherapy [
35]. Although this approach has achieved promising results in animal models, the high toxicity of IL-12 has so far hampered its clinical application.
Together with soluble factors, many different cell types harbor in the solid TME. Of note, suppressive cell populations are found both in the myeloid and in the lymphoid lineage. Regulatory T cells (Tregs), myeloid-derived suppressor cells (MDSCs), tumor-associated macrophages (TAMs), and tumor-associated neutrophils (TANs) have been extensively studied. Some of these cells, for instance TAMs, derive from an intrinsic proinflammatory and antitumorigenic macrophage phenotype, the so called M1 phenotype, characterized by Th1 cytokine secretion, but in the solid TME they convert into an anti-inflammatory and protumorigenic phenotype, the M2 phenotype, characterized by Th2 cytokine secretion [
37,
38].
The inhibitory effect of myeloid immunosuppressive cell populations on CAR T cells is currently a prominent area of research. As previously described, most immunosuppressive cells (TAMs, TANs, MDSCs) constantly release soluble factors, such as TGFb, PGE2, and IL-10, impairing CAR T cell functions [
38]. Moreover, myeloid-derived suppressive cells express on their surface the programmed cell death ligand 1 (PDL-1), which acts as an inhibitory stimulus while binding to the PD1 receptor on T cells [
39]. Based on this observation, several studies have shown that PD1 blockade improves the therapeutic efficacy of CAR T cells on solid tumors [
40]. Other groups have instead focused on depleting or re-educating the suppressor cell types.
In the lymphoid lineage, CD4+/FOXP3+ T regs are known to inhibit T cell activity at multiple stages, either via the secretion of suppressive factors (TGFb, IL-10, IL-35, adenosine), by cell-to-cell contact, or through competition for activating cytokines [
43]. Given their prominent role in generating immune tolerance and impairing T cell functions, several approaches have been attempted to deplete Tregs in the TME. However, since most Treg markers are shared with other cell populations, including CAR T cells, selective depletion of Tregs while sparing antitumor efficacy remains highly challenging [
44].
2. CAR T Cell Immune Therapy for Solid Tumors
In the last decade, the feasibility, safety, and preliminary efficacy of CAR T cells targeting a wide range of tumor antigens pertaining to solid tumors have been evaluated in early-phase trials. However, different from what has been reported for hematologic malignancies, there are several hurdles that currently limit the use of CAR T cells in the treatment of solid malignancies. When developing a CAR construct against neoplastic cells, a first key point concerns the specificity of tumor-associated antigens (TAAs), which should ideally be restricted to malignant cells and should be absent in normal cells, in order to mitigate the risk of on-target off-tumor toxicities. Another issue with TAA is their plasticity, which may lead to antigen loss or mutation, thus providing an antigen escape mechanism. The need for highly specific TAAs recognized by CAR T cells is substantiated by reports of severe toxicities caused by on-target off-tumor CAR T cells.