 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Alessandro Agnarelli | -- | 4390 | 2022-11-29 12:16:46 | | | |
| 2 | Lindsay Dong | + 2 word(s) | 4392 | 2022-11-29 12:48:41 | | |
Video Upload Options
Gastric cancer (GC) is the fifth most commonly diagnosed cancer worldwide and the fourth most common cause of cancer death. As GC is often diagnosed at an advanced stage, mortality remains very high. GC shows both genetic and environmental risk factors, with Helicobacter pylori (H. pylori) infection being the most well-described risk factor leading to GC. Germline genetic alterations are also involved in 1–3% of cases. Due to the poor survival rates of GC, immunotherapy has been widely explored as a potential treatment. Both active and passive immunotherapies have been examined. Active immunotherapies involve using a patient’s own immune system to treat the disease whereas passive immunotherapies rely on exogenous agents administered to patients such as antibodies in order to treat the tumour. The great efficacy observed in melanoma has propelled immunotherapies to be explored in a variety of other tumours, particularly breast, prostate, and lung cancer.
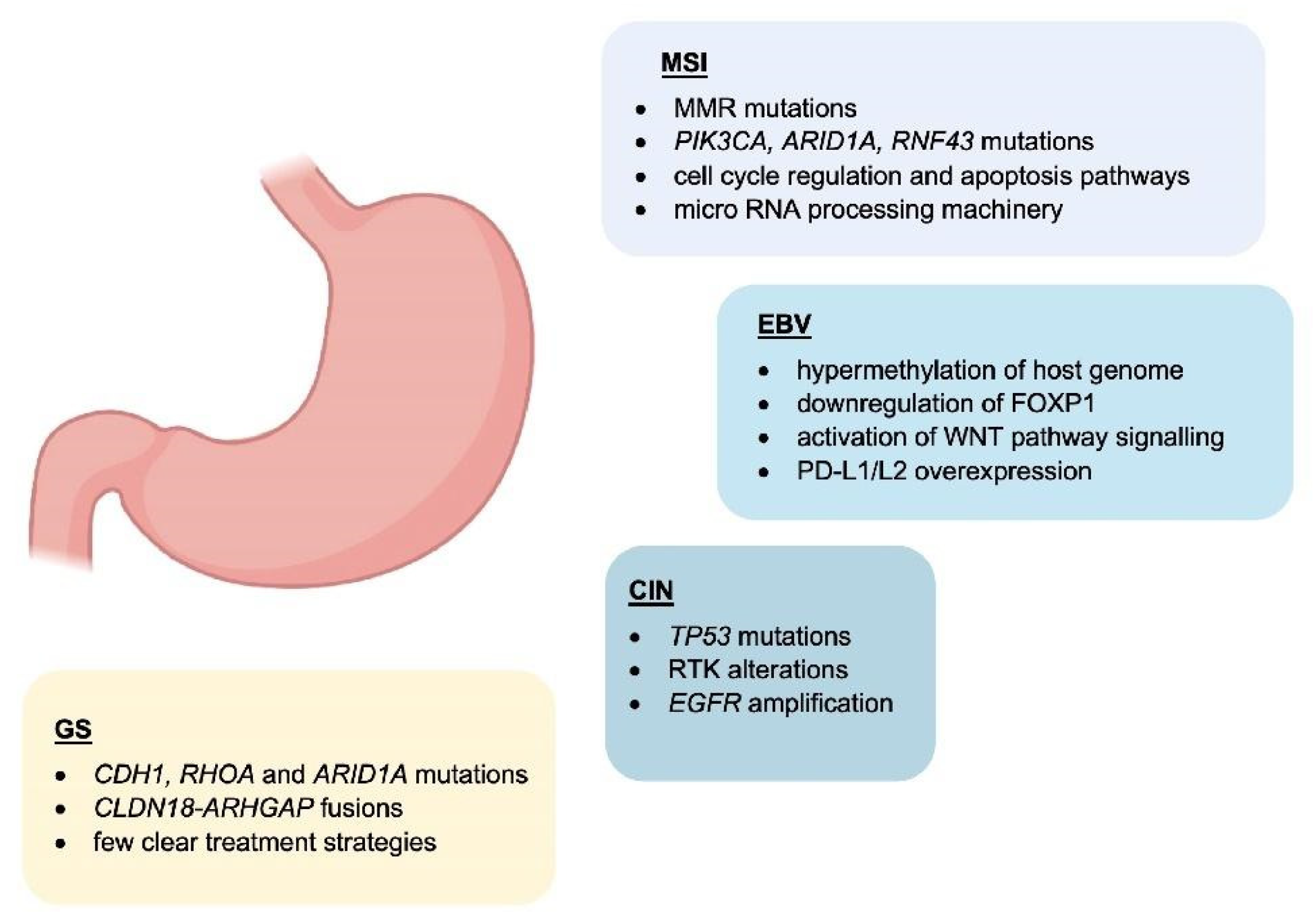
1. Molecular Classification
2. The Role of Tumour-Infiltrating Immune Cells in the Gastric Cancer Microenvironment
3. Role of Chronic Infection with Helicobacter pylori and the Association with an Immunosuppressive Microenvironment
3.1. Contribution of H. pylori Virulence Factors to Chronic Inflammation
3.2. Host Immune Response to H. pylori Infection
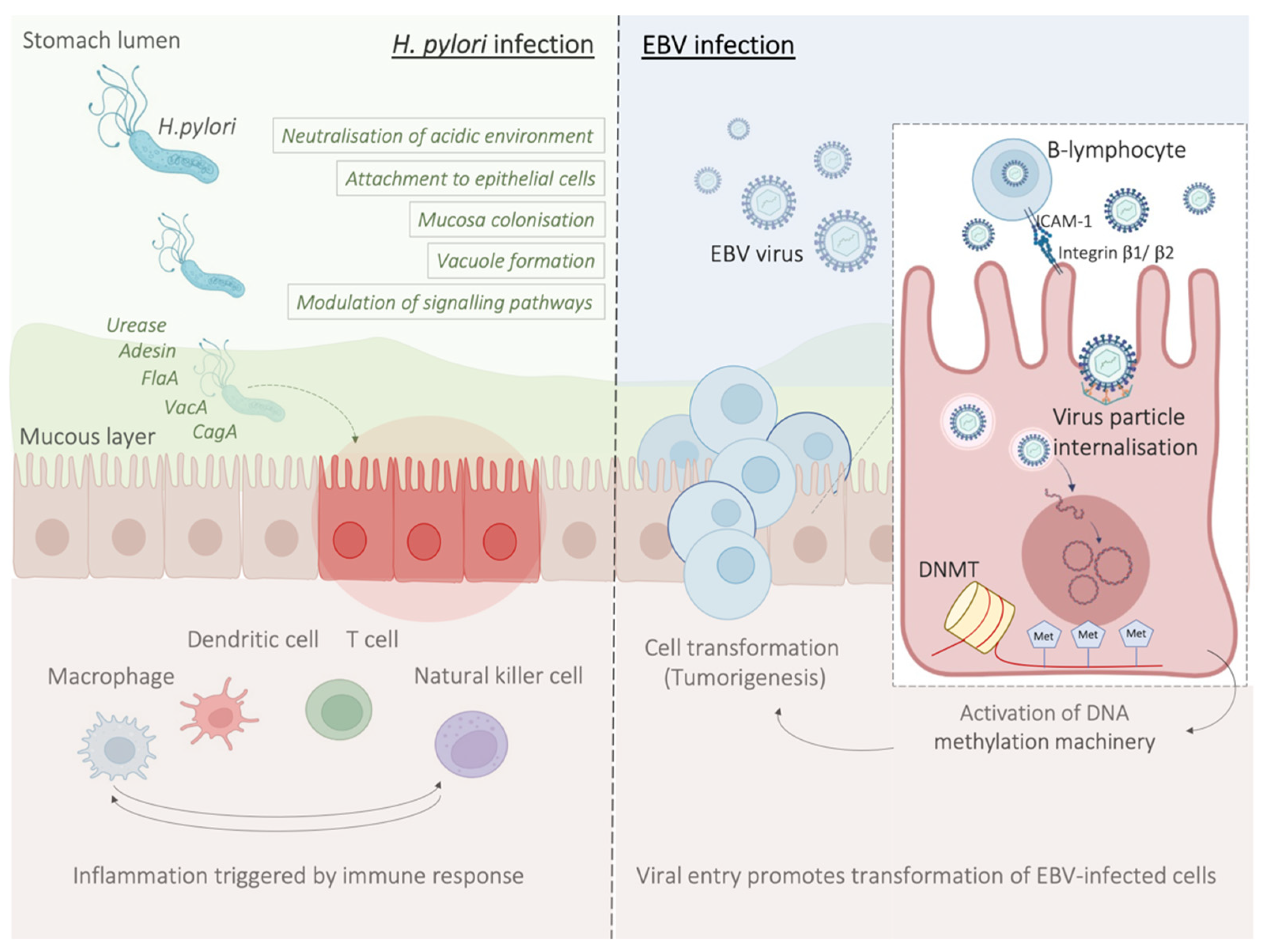
3.3. H. pylori Infection Promotes an Immunosuppressive Tumour Microenvironment
4. Molecular Mechanisms Underlying the Pathology of Microsatellite Instable Gastric Cancer
5. EBV-Related Gastric Cancer
6. Potential Role and Relevance of POLE/D Mutations in Gastric Cancer
7. HER2 Overexpression in Gastric Cancer
8. Immunotherapy in the Clinical Management of Gastric Cancer
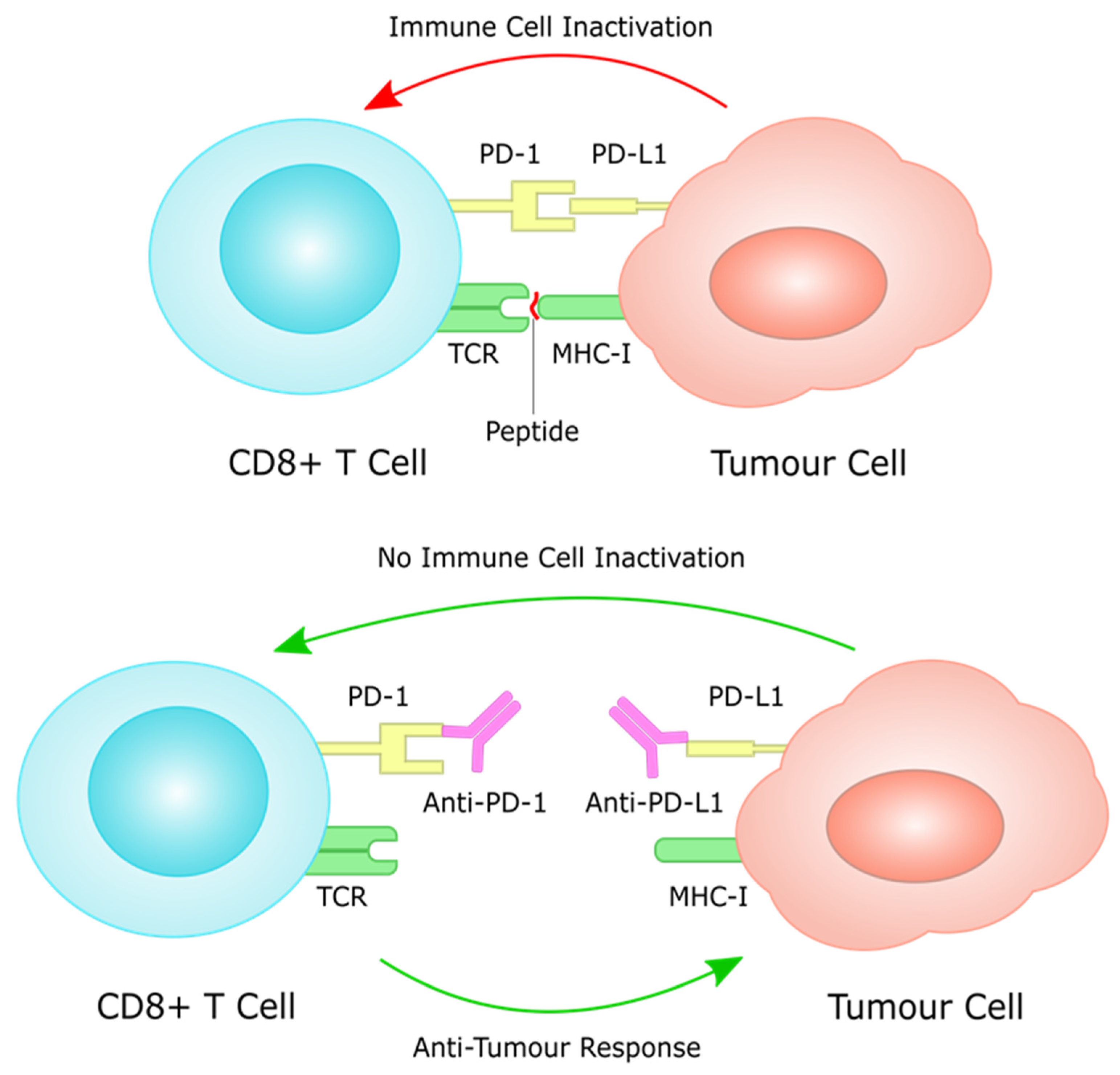
8.1. PD-L1 as a Prognostic Biomarker
8.2. Immunotherapy in Chemo-Resistant/Refractory Setting; PD-L1 Expression as a Predictive Biomarker
8.3. Immunotherapy in Previously Untreated Patients; PD-L1 Expression as a Predictive Biomarker
8.4. Immunotherapy in HER2-Positive Gastric Cancer
8.5. Immunotherapy Efficacy in Rare Subgroups: Role of MSI, EBV and TMB (Tumour Mutational Burden) Status as Predictive Biomarkers
9. Conclusions
References
- Lauren, P. The two histological main types of gastric carcinoma: Diffuse and so-called intestinal-type carcinoma. An attempt at a histo-clinical classificaTION. Acta Pathol. Microbiol. Scand. 1965, 64, 31–49.
- Milne, A.N.; Offerhaus, G.J. Early-onset gastric cancer: Learning lessons from the young. World J. Gastrointest. Oncol. 2010, 2, 59–64.
- Ma, J.; Shen, H.; Kapesa, L.; Zeng, S. Lauren classification and individualized chemotherapy in gastric cancer. Oncol. Lett. 2016, 11, 2959–2964.
- Hu, B.; El Hajj, N.; Sittler, S.; Lammert, N.; Barnes, R.; Meloni-Ehrig, A. Gastric cancer: Classification, histology and application of molecular pathology. J. Gastrointest. Oncol. 2012, 3, 251–261.
- Dicken, B.J.; Bigam, D.L.; Cass, C.; Mackey, J.R.; Joy, A.A.; Hamilton, S.M. Gastric adenocarcinoma: Review and considerations for future directions. Ann. Surg. 2005, 241, 27–39.
- Li, Z.; Wu, J.; Ji, M.; Shi, L.; Xu, B.; Jiang, J.; Wu, C. Prognostic role of lemur tyrosine kinase 3 in postoperative gastric cancer. Mol. Clin. Oncol. 2014, 2, 756–760.
- Rihawi, K.; Ricci, A.D.; Rizzo, A.; Brocchi, S.; Marasco, G.; Pastore, L.V.; Llimpe, F.L.R.; Golfieri, R.; Renzulli, M. Tumor-Associated Macrophages and Inflammatory Microenvironment in Gastric Cancer: Novel Translational Implications. Int. J. Mol. Sci. 2021, 22, 3805.
- Räihä, M.R.; Puolakkainen, P.A. Tumor-associated macrophages (TAMs) as biomarkers for gastric cancer: A review. Chronic Dis. Transl. Med. 2018, 4, 156–163.
- Wang, X.L.; Jiang, J.T.; Wu, C.P. Prognostic significance of tumor-associated macrophage infiltration in gastric cancer: A meta-analysis. Genet. Mol. Res. 2016, 15, gmr15049040.
- Zhang, Q.W.; Liu, L.; Gong, C.Y.; Shi, H.S.; Zeng, Y.H.; Wang, X.Z.; Zhao, Y.W.; Wei, Y.Q. Prognostic significance of tumor-associated macrophages in solid tumor: A meta-analysis of the literature. PLoS ONE 2012, 7, e50946.
- Wang, Z.; Yin, N.; Zhang, Z.; Zhang, Y.; Zhang, G.; Chen, W. Upregulation of T-cell Immunoglobulin and Mucin-Domain Containing-3 (Tim-3) in Monocytes/Macrophages Associates with Gastric Cancer Progression. Immunol. Investig. 2017, 46, 134–148.
- Sunakawa, Y.; Stremitzer, S.; Cao, S.; Zhang, W.; Yang, D.; Wakatsuki, T.; Ning, Y.; Yamauchi, S.; Stintzing, S.; Sebio, A.; et al. Association of variants in genes encoding for macrophage-related functions with clinical outcome in patients with locoregional gastric cancer. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2015, 26, 332–339.
- Ju, X.; Zhang, H.; Zhou, Z.; Chen, M.; Wang, Q. Tumor-associated macrophages induce PD-L1 expression in gastric cancer cells through IL-6 and TNF-ɑ signaling. Exp. Cell Res. 2020, 396, 112315.
- Shen, Z.; Kauttu, T.; Seppänen, H.; Vainionpää, S.; Ye, Y.; Wang, S.; Mustonen, H.; Puolakkainen, P. Both macrophages and hypoxia play critical role in regulating invasion of gastric cancer in vitro. Acta Oncol. 2013, 52, 852–860.
- Rahma, O.E.; Hodi, F.S. The Intersection between Tumor Angiogenesis and Immune Suppression. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2019, 25, 5449–5457.
- Harada, K.; Dong, X.; Estrella, J.S.; Correa, A.M.; Xu, Y.; Hofstetter, W.L.; Sudo, K.; Onodera, H.; Suzuki, K.; Suzuki, A.; et al. Tumor-associated macrophage infiltration is highly associated with PD-L1 expression in gastric adenocarcinoma. Gastric Cancer Off. J. Int. Gastric Cancer Assoc. Jpn. Gastric Cancer Assoc. 2018, 21, 31–40.
- Cerwenka, A.; Lanier, L.L. Natural killers join the fight against cancer. Science 2018, 359, 1460–1461.
- Martín-Antonio, B.; Suñe, G.; Perez-Amill, L.; Castella, M.; Urbano-Ispizua, A. Natural Killer Cells: Angels and Devils for Immunotherapy. Int. J. Mol. Sci. 2017, 18, 1868.
- Xia, P.; Xu, X.Y. DKK3 attenuates the cytotoxic effect of natural killer cells on CD133(+) gastric cancer cells. Mol. Carcinog. 2017, 56, 1712–1721.
- Saito, H.; Takaya, S.; Osaki, T.; Ikeguchi, M. Increased apoptosis and elevated Fas expression in circulating natural killer cells in gastric cancer patients. Gastric Cancer Off. J. Int. Gastric Cancer Assoc. Jpn. Gastric Cancer Assoc. 2013, 16, 473–479.
- González, S.; López-Soto, A.; Suarez-Alvarez, B.; López-Vázquez, A.; López-Larrea, C. NKG2D ligands: Key targets of the immune response. Trends Immunol. 2008, 29, 397–403.
- Chen, J.; Yang, J.; Jiang, J.; Zhuang, Y.; He, W. Function and subsets of dendritic cells and natural killer cells were decreased in gastric cancer. Int. J. Clin. Exp. Pathol. 2014, 7, 8304–8311.
- Li, T.; Zhang, Q.; Jiang, Y.; Yu, J.; Hu, Y.; Mou, T.; Chen, G.; Li, G. Gastric cancer cells inhibit natural killer cell proliferation and induce apoptosis via prostaglandin E2. Oncoimmunology 2016, 5, e1069936.
- Ishigami, S.; Natsugoe, S.; Tokuda, K.; Nakajo, A.; Xiangming, C.; Iwashige, H.; Aridome, K.; Hokita, S.; Aikou, T. Clinical impact of intratumoral natural killer cell and dendritic cell infiltration in gastric cancer. Cancer Lett. 2000, 159, 103–108.
- Kashimura, S.; Saze, Z.; Terashima, M.; Soeta, N.; Ohtani, S.; Osuka, F.; Kogure, M.; Gotoh, M. CD83(+) dendritic cells and Foxp3(+) regulatory T cells in primary lesions and regional lymph nodes are inversely correlated with prognosis of gastric cancer. Gastric Cancer Off. J. Int. Gastric Cancer Assoc. Jpn. Gastric Cancer Assoc. 2012, 15, 144–153.
- Kono, K.; Takahashi, A.; Sugai, H.; Fujii, H.; Choudhury, A.R.; Kiessling, R.; Matsumoto, Y. Dendritic cells pulsed with HER-2/neu-derived peptides can induce specific T-cell responses in patients with gastric cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2002, 8, 3394–3400.
- Tewari, M.; Sahai, S.; Mishra, R.R.; Shukla, S.K.; Shukla, H.S. Dendritic cell therapy in advanced gastric cancer: A promising new hope? Surg. Oncol. 2012, 21, 164–171.
- Kang, B.W.; Kim, J.G.; Lee, I.H.; Bae, H.I.; Seo, A.N. Clinical significance of tumor-infiltrating lymphocytes for gastric cancer in the era of immunology. World J. Gastrointest. Oncol. 2017, 9, 293–299.
- Kang, B.W.; Seo, A.N.; Yoon, S.; Bae, H.I.; Jeon, S.W.; Kwon, O.K.; Chung, H.Y.; Yu, W.; Kang, H.; Kim, J.G. Prognostic value of tumor-infiltrating lymphocytes in Epstein-Barr virus-associated gastric cancer. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2016, 27, 494–501.
- Grogg, K.L.; Lohse, C.M.; Pankratz, V.S.; Halling, K.C.; Smyrk, T.C. Lymphocyte-rich gastric cancer: Associations with Epstein-Barr virus, microsatellite instability, histology, and survival. Mod. Pathol. 2003, 16, 641–651.
- Lee, H.E.; Chae, S.W.; Lee, Y.J.; Kim, M.A.; Lee, H.S.; Lee, B.L.; Kim, W.H. Prognostic implications of type and density of tumour-infiltrating lymphocytes in gastric cancer. Br. J. Cancer 2008, 99, 1704–1711.
- Peek, R.M.; Blaser, M.J., Jr. Helicobacter pylori and gastrointestinal tract adenocarcinomas. Nat. Rev. Cancer 2002, 2, 28–37.
- Wroblewski, L.E.; Peek, R.M.; Wilson, K.T., Jr. Helicobacter pylori and gastric cancer: Factors that modulate disease risk. Clin. Microbiol. Rev. 2010, 23, 713–739.
- Sokic-Milutinovic, A.; Alempijevic, T.; Milosavljevic, T. Role of Helicobacter pylori infection in gastric carcinogenesis: Current knowledge and future directions. World J. Gastroenterol. 2015, 21, 11654–11672.
- Talley, N.J.; Zinsmeister, A.R.; Weaver, A.; DiMagno, E.P.; Carpenter, H.A.; Perez-Perez, G.I.; Blaser, M.J. Gastric adenocarcinoma and Helicobacter pylori infection. J. Natl. Cancer Inst. 1991, 83, 1734–1739.
- Uemura, N.; Okamoto, S.; Yamamoto, S.; Matsumura, N.; Yamaguchi, S.; Yamakido, M.; Taniyama, K.; Sasaki, N.; Schlemper, R.J. Helicobacter pylori infection and the development of gastric cancer. N. Engl. J. Med. 2001, 345, 784–789.
- Morgan, E.; Arnold, M.; Camargo, M.C.; Gini, A.; Kunzmann, A.T.; Matsuda, T.; Meheus, F.; Verhoeven, R.H.A.; Vignat, J.; Laversanne, M.; et al. The current and future incidence and mortality of gastric cancer in 185 countries, 2020–2040: A population-based modelling study. EClinicalMedicine 2022, 47, 101404.
- Alfarouk, K.O.; Bashir, A.H.H.; Aljarbou, A.N.; Ramadan, A.M.; Muddathir, A.K.; AlHoufie, S.T.S.; Hifny, A.; Elhassan, G.O.; Ibrahim, M.E.; Alqahtani, S.S.; et al. The Possible Role of Helicobacter pylori in Gastric Cancer and Its Management. Front. Oncol. 2019, 9, 75.
- Amieva, M.R.; El-Omar, E.M. Host-bacterial interactions in Helicobacter pylori infection. Gastroenterology 2008, 134, 306–323.
- Dunne, C.; Dolan, B.; Clyne, M. Factors that mediate colonization of the human stomach by Helicobacter pylori. World J. Gastroenterol. 2014, 20, 5610–5624.
- Chichirau, B.E.; Diechler, S.; Posselt, G.; Wessler, S. Tyrosine Kinases in Helicobacter pylori Infections and Gastric Cancer. Toxins 2019, 11, 591.
- Ansari, S.; Yamaoka, Y. Helicobacter pylori Virulence Factors Exploiting Gastric Colonization and its Pathogenicity. Toxins 2019, 11, 677.
- Gu, H. Role of Flagella in the Pathogenesis of Helicobacter pylori. Curr. Microbiol. 2017, 74, 863–869.
- Robinson, K.; Argent, R.H.; Atherton, J.C. The inflammatory and immune response to Helicobacter pylori infection. Best Pract. Res. Clin. Gastroenterol. 2007, 21, 237–259.
- Matos, J.I.; de Sousa, H.A.; Marcos-Pinto, R.; Dinis-Ribeiro, M. Helicobacter pylori CagA and VacA genotypes and gastric phenotype: A meta-analysis. Eur. J. Gastroenterol. Hepatol. 2013, 25, 1431–1441.
- Fuccio, L.; Eusebi, L.H.; Bazzoli, F. Gastric cancer, Helicobacter pylori infection and other risk factors. World J. Gastrointest. Oncol. 2010, 2, 342–347.
- Karkhah, A.; Ebrahimpour, S.; Rostamtabar, M.; Koppolu, V.; Darvish, S.; Vasigala, V.K.R.; Validi, M.; Nouri, H.R. Helicobacter pylori evasion strategies of the host innate and adaptive immune responses to survive and develop gastrointestinal diseases. Microbiol. Res. 2019, 218, 49–57.
- Müller, A.; Oertli, M.; Arnold, I.C.H. H. pylori exploits and manipulates innate and adaptive immune cell signaling pathways to establish persistent infection. Cell Commun. Signal. 2011, 9, 25.
- Lima de Souza Gonçalves, V.; Cordeiro Santos, M.L.; Silva Luz, M.; Santos Marques, H.; de Brito, B.B.; França da Silva, F.A.; Souza, C.L.; Oliveira, M.V.; de Melo, F.F. From Helicobacter pylori infection to gastric cancer: Current evidence on the immune response. World J. Clin. Oncol. 2022, 13, 186–199.
- Wilson, K.T.; Crabtree, J.E. Immunology of Helicobacter pylori: Insights into the failure of the immune response and perspectives on vaccine studies. Gastroenterology 2007, 133, 288–308.
- Oster, P.; Vaillant, L.; McMillan, B.; Velin, D. The Efficacy of Cancer Immunotherapies Is Compromised by Helicobacter pylori Infection. Front. Immunol. 2022, 13, 899161.
- Oster, P.; Vaillant, L.; Riva, E.; McMillan, B.; Begka, C.; Truntzer, C.; Richard, C.; Leblond, M.M.; Messaoudene, M.; Machremi, E.; et al. Helicobacter pylori infection has a detrimental impact on the efficacy of cancer immunotherapies. Gut 2022, 71, 457–466.
- Liu, M.; Guo, F. Recent updates on cancer immunotherapy. Precis. Clin. Med. 2018, 1, 65–74.
- Paluch, C.; Santos, A.M.; Anzilotti, C.; Cornall, R.J.; Davis, S.J. Immune Checkpoints as Therapeutic Targets in Autoimmunity. Front. Immunol. 2018, 9, 2306.
- Lengauer, C.; Kinzler, K.W.; Vogelstein, B. Genetic instabilities in human cancers. Nature 1998, 396, 643–649.
- Belien, J.A.M.; Buffart, T.E.; Gill, A.J.; Broeckaert, M.A.M.; Quirke, P.; Meijer, G.A.; Grabsch, H.I. Gross genomic damage measured by DNA image cytometry independently predicts gastric cancer patient survival. Br. J. Cancer 2009, 101, 1011–1018.
- Ratti, M.; Lampis, A.; Hahne, J.C.; Passalacqua, R.; Valeri, N. Microsatellite instability in gastric cancer: Molecular bases, clinical perspectives, and new treatment approaches. Cell. Mol. Life Sci. 2018, 75, 4151–4162.
- Puliga, E.; Corso, S.; Pietrantonio, F.; Giordano, S. Microsatellite instability in Gastric Cancer: Between lights and shadows. Cancer Treat. Rev. 2021, 95, 102175.
- Julié, C.; Trésallet, C.; Brouquet, A.; Vallot, C.; Zimmermann, U.; Mitry, E.; Radvanyi, F.; Rouleau, E.; Lidereau, R.; Coulet, F.; et al. Identification in daily practice of patients with Lynch syndrome (hereditary nonpolyposis colorectal cancer): Revised Bethesda guidelines-based approach versus molecular screening. Am. J. Gastroenterol. 2008, 103, 2825–2835, quiz 36.
- Hudler, P. Genetic aspects of gastric cancer instability. Sci. World J. 2012, 2012, 761909.
- Yuza, K.; Nagahashi, M.; Watanabe, S.; Takabe, K.; Wakai, T. Hypermutation and microsatellite instability in gastrointestinal cancers. Oncotarget 2017, 8, 112103–112115.
- Leite, M.; Corso, G.; Sousa, S.; Milanezi, F.; Afonso, L.P.; Henrique, R.; Soares, J.M.; Castedo, S.; Carneiro, F.; Roviello, F.; et al. MSI phenotype and MMR alterations in familial and sporadic gastric cancer. Int. J. Cancer 2011, 128, 1606–1613.
- Gu, M.; Kim, D.; Bae, Y.; Choi, J.; Kim, S.; Song, S. Analysis of microsatellite instability, protein expression and methylation status of hMLH1 and hMSH2 genes in gastric carcinomas. Hepato-Gastroenterology 2009, 56, 899–904.
- Bevilacqua, R.A.; Simpson, A.J. Methylation of the hMLH1 promoter but no hMLH1 mutations in sporadic gastric carcinomas with high-level microsatellite instability. Int. J. Cancer 2000, 87, 200–203.
- Wu, M.-S.; Sheu, J.-C.; Shun, C.-T.; Lee, W.-J.; Wang, J.-T.; Wang, T.-H.; Cheng, A.-L.; Lin, J.-T. Infrequent hMSH2 mutations in sporadic gastric adenocarcinoma with microsatellite instability. Cancer Lett. 1997, 112, 161–166.
- Sugimoto, R.; Sugai, T.; Habano, W.; Endoh, M.; Eizuka, M.; Yamamoto, E.; Uesugi, N.; Ishida, K.; Kawasaki, T.; Matsumoto, T.; et al. Clinicopathological and molecular alterations in early gastric cancers with the microsatellite instability-high phenotype. Int. J. Cancer 2016, 138, 1689–1697.
- Li, B.; Liu, H.Y.; Guo, S.H.; Sun, P.; Gong, F.M.; Jia, B.Q. Microsatellite instability of gastric cancer and precancerous lesions. Int. J. Clin. Exp. Med. 2015, 8, 21138–21144.
- Ling, Z.-Q.; Tanaka, A.; Li, P.; Nakayama, T.; Fujiyama, Y.; Hattori, T.; Sugihara, H. Microsatellite instability with promoter methylation and silencing of hMLH1 can regionally occur during progression of gastric carcinoma. Cancer Lett. 2010, 297, 244–251.
- Corso, G.; Velho, S.; Paredes, J.; Pedrazzani, C.; Martins, D.; Milanezi, F.; Pascale, V.; Vindigni, C.; Pinheiro, H.; Leite, M.; et al. Oncogenic mutations in gastric cancer with microsatellite instability. Eur. J. Cancer 2011, 47, 443–451.
- Ligtenberg, M.J.; Kuiper, R.P.; Chan, T.L.; Goossens, M.; Hebeda, K.M.; Voorendt, M.; Lee, T.Y.; Bodmer, D.; Hoenselaar, E.; Hendriks-Cornelissen, S.J.; et al. Heritable somatic methylation and inactivation of MSH2 in families with Lynch syndrome due to deletion of the 3′ exons of TACSTD1. Nat. Genet. 2009, 41, 112–117.
- Kovacs, M.E.; Papp, J.; Szentirmay, Z.; Otto, S.; Olah, E. Deletions removing the last exon of TACSTD1 constitute a distinct class of mutations predisposing to Lynch syndrome. Hum. Mutat. 2009, 30, 197–203.
- Takayama, T.; Sakabe, T.; Fujii, M.; Yamada, E.; Uno, M.; Ono, Y. In vitro production of human antibodies specifically reactive with human gastric cancer cells of established lines and autologous tissues. J. Surg. Oncol. 1987, 36, 215–224.
- Bocian, J.; Januszkiewicz-Lewandowska, D. Epstein-Barr virus infection—Life cycle, methods of diagnosis, associated diseases. Postepy Hig. I Med. Dosw. 2011, 65, 286–298.
- Rodriquenz, M.G.; Roviello, G.; D′Angelo, A.; Lavacchi, D.; Roviello, F.; Polom, K. MSI and EBV Positive Gastric Cancer’s Subgroups and Their Link with Novel Immunotherapy. J. Clin. Med. 2020, 9, 1427.
- Sun, K.; Jia, K.; Lv, H.; Wang, S.Q.; Wu, Y.; Lei, H.; Chen, X. EBV-Positive Gastric Cancer: Current Knowledge and Future Perspectives. Front. Oncol. 2020, 10, 583463.
- Lieberman, P.M. Virology. Epstein-Barr virus turns 50. Science 2014, 343, 1323–1325.
- Hutt-Fletcher, L.M. The Long and Complicated Relationship between Epstein-Barr Virus and Epithelial Cells. J. Virol. 2016, 91, e01677-16.
- Nanbo, A.; Kachi, K.; Yoshiyama, H.; Ohba, Y. Epstein-Barr virus exploits host endocytic machinery for cell-to-cell viral transmission rather than a virological synapse. J. Gen. Virol. 2016, 97, 2989–3006.
- Lieberman, P.M. Keeping it quiet: Chromatin control of gammaherpesvirus latency. Nat. Rev. Microbiol. 2013, 11, 863–875.
- Rowlands, D.C.; Ito, M.; Mangham, D.C.; Reynolds, G.; Herbst, H.; Hallissey, M.T.; Fielding, J.W.; Newbold, K.M.; Jones, E.L.; Young, L.S.; et al. Epstein-Barr virus and carcinomas: Rare association of the virus with gastric adenocarcinomas. Br. J. Cancer 1993, 68, 1014–1019.
- Shinozaki, A.; Sakatani, T.; Ushiku, T.; Hino, R.; Isogai, M.; Ishikawa, S.; Uozaki, H.; Takada, K.; Fukayama, M. Downregulation of microRNA-200 in EBV-associated gastric carcinoma. Cancer Res. 2010, 70, 4719–4727.
- Zheng, X.; Wang, J.; Wei, L.; Peng, Q.; Gao, Y.; Fu, Y.; Lu, Y.; Qin, Z.; Zhang, X.; Lu, J.; et al. Epstein-Barr Virus MicroRNA miR-BART5-3p Inhibits p53 Expression. J. Virol. 2018, 92, e01022-18.
- Song, Y.; Li, Q.; Liao, S.; Zhong, K.; Jin, Y.; Zeng, T. Epstein-Barr virus-encoded miR-BART11 promotes tumor-associated macrophage-induced epithelial-mesenchymal transition via targeting FOXP1 in gastric cancer. Virology 2020, 548, 6–16.
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520.
- Liu, J.; Zhou, M.; Ouyang, Y.; Du, L.; Xu, L.; Li, H. Identification of potential biomarkers and their clinical significance in gastric cancer using bioinformatics analysis methods. PeerJ 2020, 8, e9174.
- Strosberg, J.; Mizuno, N.; Doi, T.; Grande, E.; Delord, J.P.; Shapira-Frommer, R.; Bergsland, E.; Shah, M.; Fakih, M.; Takahashi, S.; et al. Efficacy and Safety of Pembrolizumab in Previously Treated Advanced Neuroendocrine Tumors: Results from the Phase II KEYNOTE-158 Study. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2020, 26, 2124–2130.
- Kono, K.; Nakajima, S.; Mimura, K. Current status of immune checkpoint inhibitors for gastric cancer. Gastric Cancer Off. J. Int. Gastric Cancer Assoc. Jpn. Gastric Cancer Assoc. 2020, 23, 565–578.
- Sundar, R.; Smyth, E.C.; Peng, S.; Yeong, J.P.S.; Tan, P. Predictive Biomarkers of Immune Checkpoint Inhibition in Gastroesophageal Cancers. Front. Oncol. 2020, 10, 763.
- Burgers, P.M.J.; Kunkel, T.A. Eukaryotic DNA Replication Fork. Annu. Rev. Biochem. 2017, 86, 417–438.
- Chalmers, Z.R.; Connelly, C.F.; Fabrizio, D.; Gay, L.; Ali, S.M.; Ennis, R.; Schrock, A.; Campbell, B.; Shlien, A.; Chmielecki, J.; et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 2017, 9, 34.
- Wang, F.; Zhao, Q.; Wang, Y.N.; Jin, Y.; He, M.M.; Liu, Z.X.; Xu, R.H. Evaluation of POLE and POLD1 Mutations as Biomarkers for Immunotherapy Outcomes Across Multiple Cancer Types. JAMA Oncol. 2019, 5, 1504–1506.
- Abrahao-Machado, L.F.; Scapulatempo-Neto, C. HER2 testing in gastric cancer: An update. World J. Gastroenterol. 2016, 22, 4619–4625.
- Baykara, M.; Benekli, M.; Ekinci, O.; Irkkan, S.C.; Karaca, H.; Demirci, U.; Akinci, M.B.; Unal, O.U.; Dane, F.; Turkoz, F.P.; et al. Clinical Significance of HER2 Overexpression in Gastric and Gastroesophageal Junction Cancers. J. Gastrointest. Surg. 2015, 19, 1565–1571.
- Jørgensen, J.T.; Hersom, M. HER2 as a Prognostic Marker in Gastric Cancer—A Systematic Analysis of Data from the Literature. J. Cancer 2012, 3, 137–144.
- Bang, Y.J.; Van Cutsem, E.; Feyereislova, A.; Chung, H.C.; Shen, L.; Sawaki, A.; Lordick, F.; Ohtsu, A.; Omuro, Y.; Satoh, T.; et al. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): A phase 3, open-label, randomised controlled trial. Lancet 2010, 376, 687–697.
- Whiteside, T.L. The tumor microenvironment and its role in promoting tumor growth. Oncogene 2008, 27, 5904–5912.
- Suh, K.J.; Sung, J.H.; Kim, J.W.; Han, S.-H.; Lee, H.S.; Min, A.; Kang, M.H.; Kim, J.E.; Kim, J.-W.; Kim, S.H.; et al. EGFR or HER2 inhibition modulates the tumor microenvironment by suppression of PD-L1 and cytokines release. Oncotarget 2017, 8, 63901–63910.
- Morihiro, T.; Kuroda, S.; Kanaya, N.; Kakiuchi, Y.; Kubota, T.; Aoyama, K.; Tanaka, T.; Kikuchi, S.; Nagasaka, T.; Nishizaki, M.; et al. PD-L1 expression combined with microsatellite instability/CD8+ tumor infiltrating lymphocytes as a useful prognostic biomarker in gastric cancer. Sci. Rep. 2019, 9, 4633.
- Chang, H.; Jung, W.Y.; Kang, Y.; Lee, H.; Kim, A.; Kim, H.K.; Shin, B.K.; Kim, B.H. Programmed death-ligand 1 expression in gastric adenocarcinoma is a poor prognostic factor in a high CD8+ tumor infiltrating lymphocytes group. Oncotarget 2016, 7, 80426–80434.
- Kang, Y.K.; Boku, N.; Satoh, T.; Ryu, M.H.; Chao, Y.; Kato, K.; Chung, H.C.; Chen, J.S.; Muro, K.; Kang, W.K.; et al. Nivolumab in patients with advanced gastric or gastro-oesophageal junction cancer refractory to, or intolerant of, at least two previous chemotherapy regimens (ONO-4538-12, ATTRACTION-2): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 390, 2461–2471.
- Chen, L.T.; Satoh, T.; Ryu, M.H.; Chao, Y.; Kato, K.; Chung, H.C.; Chen, J.S.; Muro, K.; Kang, W.K.; Yeh, K.H.; et al. A phase 3 study of nivolumab in previously treated advanced gastric or gastroesophageal junction cancer (ATTRACTION-2): 2-year update data. Gastric Cancer Off. J. Int. Gastric Cancer Assoc. Jpn. Gastric Cancer Assoc. 2020, 23, 510–519.
- Shitara, K.; Van Cutsem, E.; Bang, Y.J.; Fuchs, C.; Wyrwicz, L.; Lee, K.W.; Kudaba, I.; Garrido, M.; Chung, H.C.; Lee, J.; et al. Efficacy and Safety of Pembrolizumab or Pembrolizumab Plus Chemotherapy vs Chemotherapy Alone for Patients with First-line, Advanced Gastric Cancer: The KEYNOTE-062 Phase 3 Randomized Clinical Trial. JAMA Oncol. 2020, 6, 1571–1580.
- Wainberg, Z.A.; Shitara, K.; Van Cutsem, E.; Wyrwicz, L.; Lee, K.W.; Kudaba, I.; Garrido, M.; Chung, H.C.C.; Lee, J.; Castro-Salguero, H.R.; et al. Pembrolizumab with or without chemotherapy versus chemotherapy alone for patients with PD-L1–positive advanced gastric or gastroesophageal junction adenocarcinoma: Update from the phase 3 KEYNOTE-062 trial. J. Clin. Oncol. 2022, 40 (Suppl. 4), iv152–iv153.
- Janjigian, Y.Y.; Kawazoe, A.; Yañez, P.; Li, N.; Lonardi, S.; Kolesnik, O.; Barajas, O.; Bai, Y.; Shen, L.; Tang, Y.; et al. The KEYNOTE-811 trial of dual PD-1 and HER2 blockade in HER2-positive gastric cancer. Nature 2021, 600, 727–730.
- Kawazoe, A.; Shitara, K.; Kuboki, Y.; Bando, H.; Kojima, T.; Yoshino, T.; Ohtsu, A.; Ochiai, A.; Togashi, Y.; Nishikawa, H.; et al. Clinicopathological features of 22C3 PD-L1 expression with mismatch repair, Epstein-Barr virus status, and cancer genome alterations in metastatic gastric cancer. Gastric Cancer Off. J. Int. Gastric Cancer Assoc. Jpn. Gastric Cancer Assoc. 2019, 22, 69–76.
- Marabelle, A.; Le, D.T.; Ascierto, P.A.; Di Giacomo, A.M.; De Jesus-Acosta, A.; Delord, J.P.; Geva, R.; Gottfried, M.; Penel, N.; Hansen, A.R.; et al. Efficacy of Pembrolizumab in Patients with Noncolorectal High Microsatellite Instability/Mismatch Repair-Deficient Cancer: Results From the Phase II KEYNOTE-158 Study. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2020, 38, 1–10.
- Chao, J.; Fuchs, C.S.; Shitara, K.; Tabernero, J.; Muro, K.; Van Cutsem, E.; Bang, Y.J.; De Vita, F.; Landers, G.; Yen, C.J.; et al. Assessment of Pembrolizumab Therapy for the Treatment of Microsatellite Instability-High Gastric or Gastroesophageal Junction Cancer Among Patients in the KEYNOTE-059, KEYNOTE-061, and KEYNOTE-062 Clinical Trials. JAMA Oncol. 2021, 7, 895–902.
- Kim, S.T.; Cristescu, R.; Bass, A.J.; Kim, K.M.; Odegaard, J.I.; Kim, K.; Liu, X.Q.; Sher, X.; Jung, H.; Lee, M.; et al. Comprehensive molecular characterization of clinical responses to PD-1 inhibition in metastatic gastric cancer. Nat. Med. 2018, 24, 1449–1458.

