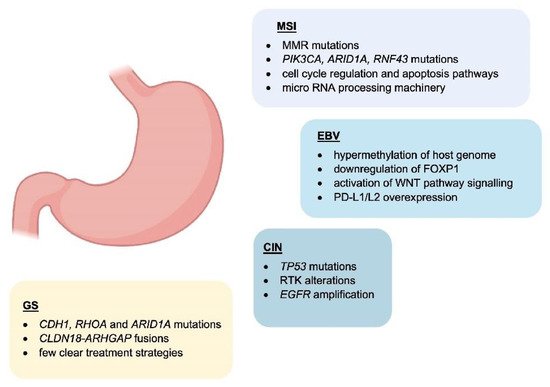
Epstein–Barr virus (EBV) emerged as an important virulence factor for nasopharyngeal carcinoma. EBV infection also causes the development of T-cell lymphoma and EBV-associated GC (EBVaGC) [
106,
107]. Immunotherapy drug treatments were successful against EBV-positive and MSI GCs [
108]. Once EBV infects the human body, this does not immediately produce GC and EBV-positive GC is not characterized by any evident clinical manifestations [
109,
110]. Two theories have been reported about the mechanism of EBV infection. According to the first theory, EBV produces the infection of B-lymphocytes and oral epithelial cells [
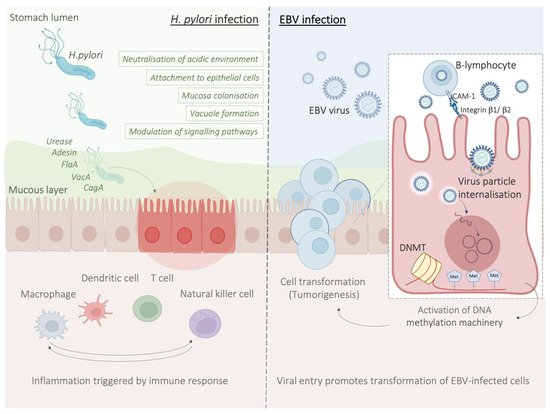
111]. In particular, since EBV is present in the saliva, this causes the infection of the epithelial cells [
111]. The second theory reports EBV reactivation in B-lymphocytes in the stomach and its subsequent release to cause the infection of epithelial cells [
111]. EBV infection of lymphocytes results in the interaction of these cells with epithelial cells [
112]. This interaction is mediated by integrin β-1/β-2 and the translocation of intracellular adhesion molecule-1 to the cell surface (ICAM-1) produce cell-to cell contact [
112]. The virus particles are then internalised by recipient cells through clathrin-mediated endocytosis [
112]. EBV-particles inside the host cell nucleus are characterized by a naked, linear DNA genomes and a specific protein capsid protect them [
113]. The exposed DNA linear genome is then circularised into a functional chromosome [
113]. After circularisation, the chromatinised viral DNA protects it from DNA damage and provides accurate regulation of gene expression [
113] (
Figure 2). EBV genome is characterised by widely methylated CpG motifs enabling it to establish latent infection [
114]. The two types of infection caused by EBV are the lytic and the latent form [
114]. The latent infection is the one preferred by EBV and during the long incubation period, EBV causes the methylation of the host DNA and the expansion of GC [
113,
114]. Latent EBV proteins such as EBERs, BARF-0, EBNA-1, and LMP2A downregulate the miR-200 family causing a reduction in E-cadherin expression [
115]. This mechanism is mediated by the upregulation of the E-cadherin repressors, ZEB1 and ZEB2 [
115]. Tumour progression involves the loss of cell-to-cell adhesion and this event is also an important step in the carcinogenesis of EBVaGC [
115]. EBV is the first human virus expressing many microRNAs [
116]. The EBV miRNA BART11 has been shown to downregulate forkhead box protein P1 (FOXP1) transcription factor [
117]. FOXP1 downregulation activates the epithelial-mesenchymal transition involving the gastric tumour cells or affecting the tumour microenvironment [
117]. This, in turn, accelerates cancer invasion and metastasis, thereby affecting the survival and prognosis of patients [
117].
6. Potential Role and Relevance of POLE/D Mutations in Gastric Cancer
The immune checkpoint inhibitor (ICI) biomarkers approved by the US Food and Drug Administration (FDA) in the treatment of certain cancers are the use of PD-L1 expression, microsatellite instability (MSI)-H/deficient mismatch repair (dMMR) and tumoural mutation burden (TMB) [
123,
124,
125]. Since some responses seen with ICIs do not fully correlate with any of the biomarkers above, other potential predictive biomarkers of response to ICI can exist in GC [
126,
127].
POLE and
POLD1 genes encode for the DNA polymerases ε and δ [
127,
128]. TMB and clinical benefits of immunotherapy have been associated with mutations in
POLE and
POLD1 in different cancer types [
129,
130].
7. HER2 Overexpression in Gastric Cancer
Human epidermal growth factor receptor 2 (HER2) is a receptor tyrosine kinase proto-oncogene that is increasingly understood to be overexpressed in GC. Different studies have found HER2 overexpression to occur in between 4.4% and 53.4% of GCs, with an average of 17.9% [
132]. Prognostically, HER2 appears to be correlated with poorer survival and increased recurrence in GC [
133,
134]. HER2 targeting through the monoclonal antibody trastuzumab together with chemotherapy has in fact been shown to increase survival in HER2+ GC patients; however, the effect is small with a clinical trial only finding survival increasing from 11.1 months to 13.8 months [
135].
During treatment, the tumour microenvironment contributes to tumourigenesis and proliferation but is also associated with immune cell infiltration and treatment efficacy [
136]. Suh et al. reported that the HER2 pathway modulates the tumour microenvironment and this is correlated with tumour pathological characteristics and patient survival [
137].
8. Immunotherapy in the Clinical Management of Gastric Cancer
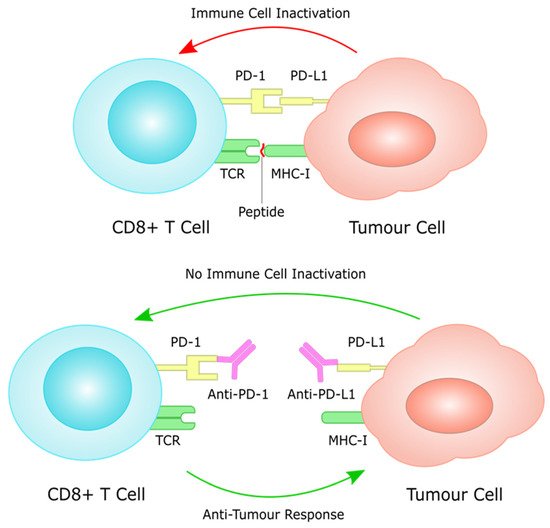
Over the last few years, immunotherapy has been actively incorporated as part of first and later lines of systemic treatment for advanced gastric cancer. PD-1 inhibitors have been shown to significantly improve efficacy in several large phase III trials when added to platinum-based chemotherapy, which has for many years been the standard of care in the first line setting for metastatic GC (Figure 3).
Figure 3. PD-L1 on tumour cells works through the PD-1 receptor on T cells to induce immune cell inactivation. Treatment with immune checkpoint inhibitors inhibits this interaction, often through monoclonal antibodies against PD-1 and PD-L1, restoring immune cell function against tumour cells.
8.1. PD-L1 as a Prognostic Biomarker
There have been several studies investigating a potential prognostic role for PD-L1 expression in GC, with diverse and controversial results. In a retrospective study by Morihiro, et al., including 283 patients with GC, PD-L1 expression was significantly correlated with a poor prognosis (
p = 0.0025). Multivariate analysis revealed that PD-L1 expression was found to be an independent poor prognostic factor, along with diffuse histological type and lymph node involvement. [
140]. Furthermore, Chang et al., using tissue microarrays in 464 GC samples, showed that PD-L1 and PD-1 expression was significantly correlated with several adverse prognostic pathologic features, such as T stage, lymphatic invasion and diffuse Lauren histologic type. In the same study, subgroup analyses in which patients were divided into two groups according to CD8+ TILs expression levels (high and low), it was shown that high PD-L1 expression was a negative prognostic factor only in the high CD8+ TILs subgroup [
141].
8.2. Immunotherapy in Chemo-Resistant/Refractory Setting; PD-L1 Expression as a Predictive Biomarker
One of the first phase III trials that investigated the use of PD-1 monotherapy in the chemo-refractory setting in metastatic GC was the ATTRACTION-2 trial, which randomised previously treated patients with metastatic gastric or gastro–oesophageal junction (GOJ) adenocarcinoma with more than two lines of treatment, to nivolumab or placebo. Median overall survival was 5.26 months (95% CI 4.60–6.37) in the nivolumab group and 4.14 months (3.42–4.86) in the placebo group (hazard ratio 0.63, 95% CI 0.51–0.78;
p < 0.0001). [
147]. A more recent update of the trial with 2-year follow-up data confirmed the long-term benefit of nivolumab over placebo, with a higher OS rate noted in the nivolumab vs. placebo group at 2 years (10.6% vs. 3.2%). The OS benefit was observed regardless of tumour PD-L1 expression, although this must be interpreted with caution because only 192 of 493 patients (39%) had PD-L1 results available for analysis [
148]. Despite the fact that the main population of the study was Asian patients, the positive benefit of nivolumab in overall survival established this option as a standard third line and beyond option regardless of PD-L1 expression levels in many countries.
8.3. Immunotherapy in Previously Untreated Patients; PD-L1 Expression as a Predictive Biomarker
The next logical step was to investigate the incorporation of immunotherapy in the first line setting, in previously untreated patients, with advanced gastric cancer. One of the first phase III trials that attempted to answer this specific question was the Keynote-062 trial. In this trial, patients were randomized 1:1:1 to pembrolizumab, pembrolizumab plus chemotherapy (cisplatin plus fluorouracil or capecitabine) or chemotherapy plus placebo. Primary end points were OS and PFS in patients with PD-L1 CPS ≥ 1 or ≥10. This trial demonstrated non-inferiority for pembrolizumab to chemotherapy for OS in patients with CPS ≥ 1 (median, 10.6 vs. 11.1 months; hazard ratio [HR], 0.91; 99.2% CI, 0.69–1.18), however pembrolizumab monotherapy was not found to be superior to chemotherapy in patients with CPS ≥ 1. In patients with CPS ≥ 10, pembrolizumab numerically prolonged OS vs. chemotherapy (median, 17.4 vs. 10.8 months; HR, 0.69; 95% CI, 0.49–0.97); however, this difference was not tested for statistical significance. Pembrolizumab combination with chemotherapy was not found to be superior to chemotherapy for OS in patients with CPS ≥ 1 (12.5 vs. 11.1 months; HR, 0.85; 95% CI, 0.70–1.03;
p = 0.05) or CPS ≥ 10 (12.3 vs. 10.8 months; HR, 0.85; 95% CI, 0.62–1.17;
p = 0.16) or for PFS in patients with CPS ≥ 1 (6.9 vs. 6.4 months; HR, 0.84; 95% CI, 0.70–1.02;
p = 0.04) [
152]. In a more recent update of the trial in ASCO GI 2022, with a median follow-up of 54.3 months (range, 46.8–66.1), efficacy was consistent with the initial final analysis data; 24-month OS rates (pembrolizumab vs. chemotherapy) were 26.6% versus 18.8% in the CPS ≥ 1 population and 39.1% versus 21.1% in the CPS ≥ 10 population. Twenty-four-month OS rates (pembrolizumab + chemotherapy vs. chemotherapy) were 24.5% versus 18.8% in the CPS ≥ 1 population and 28.3% versus 21.1% in the CPS ≥ 10 population [
153]. Although Keynote-062 was considered a negative trial, it provided further valuable supporting evidence with regards to the potential predictive role of PD-L1 CPS in terms of the selection of patients that would most benefit from immunotherapy, namely patients with PD-L1 CPS ≥ 1, and ideally PD-L1 CPS ≥ 10.
8.4. Immunotherapy in HER2-Positive Gastric Cancer
PD-1 inhibition is thought to work synergistically with HER2 inhibition increasing ADCC (antibody-dependent cellular cytotoxicity), and this concept has been investigated in the Keynote-811 trial, where treatment naive patients with HER2-positive GC or GOJ adenocarcinoma were randomized to receive pembrolizumab or placebo plus trastuzumab and investigator’s choice of fluorouracil/cisplatin or capecitabine/oxaliplatin. Overall, 84% of patients had a PD-L1 CPS ≥ 1. Confirmed ORR (95% CI) was 74.4% (66.2–81.6) for pembrolizumab + chemotherapy vs. 51.9% (43.0–60.7) for placebo + chemotherapy,
p = 0.00006); CR (complete response) rate was 11.3% vs. 3.1% and DCR (disease control rate) (95% CI) was 96.2% (91.4–98.8) vs. 89.3 (82.7–94.0). [
160]. The very encouraging preliminary results from the Keynote-811 provide a proof of concept for the synergistic effect of PD-1 and HER2 inhibition in HER2-positive gastric cancer, which stands a very good chance to consist of a new standard of care in the first line setting, providing long-term analysis also reveals a survival benefit for pembrolizumab, trastuzumab and chemotherapy in this setting.
8.5. Immunotherapy Efficacy in Rare Subgroups: Role of MSI, EBV and TMB (Tumour Mutational Burden) Status as Predictive Biomarkers
The role of immunotherapy in the management of mismatch-repair deficient (MMR-D) and EBV-associated gastric tumours has long been established through several studies. In stage IV advanced gastric cancer, EBV-positive and MMR-D tumours are identified both in approximately 6% of cases [
161]. In the Keynote-158 phase II trial investigating the efficacy of pembrolizumab in MMR-D tumours, 11 of 24 patients with advanced gastric cancer demonstrated an objective response as their best response (ORR, 45.8%), which was associated with a median PFS of 11.0 months and a median OS which was not reached [
162]. In a post hoc analysis of the outcomes of MSI-H GC patients included in the phase 2 single-arm trial KEYNOTE-059 (≥3 lines of treatment) and the phase 3 KEYNOTE-061 (second line) and KEYNOTE-062 (first line treatment) trials, the median overall survival was not reached (NR) for pembrolizumab in all three of these trials. The median PFS) for pembrolizumab was NR (95% CI, 1.1 months to NR) in KEYNOTE-059 and 17.8 months (95% CI, 2.7 months to NR) in KEYNOTE-061 (vs. 3.5 months for chemotherapy). In KEYNOTE-062, the median PFS was 11.2 months (95% CI, 1.5 months to NR) for pembrolizumab, NR (95% CI, 3.6 months to NR) for pembrolizumab plus chemotherapy, and only 6.6 months (95% CI, 4.4–8.3 months) for chemotherapy alone. The ORR for pembrolizumab was 57.1% in KEYNOTE-059 and 46.7% (vs 16.7% for chemotherapy alone) in KEYNOTE-061. In KEYNOTE-062, the ORR was 57.1% for pembrolizumab, 64.7% for pembrolizumab plus chemotherapy, and 36.8% for chemotherapy alone [
163]. Very few clinical trial data exist in regards to the efficacy of immunotherapy in EBV-positive GC; in a phase II study of pembrolizumab in metastatic GC patients with MSI-H and Epstein–Barr virus-positive tumours, impressive responses to pembrolizumab were noted (overall response rate (ORR) 85.7% in MSI-H metastatic GC and ORR 100% in Epstein–Barr virus-positive tumours) [
164]. The results above unequivocally establish MSI-H status and EBV positivity as by far the most robust biomarkers of response to immunotherapy in GC, however it has to be noted that despite the high and durable responses, there is a significant fraction of patients which test positive for the above biomarkers that do not respond to immunotherapy, and therefore there is a great unmet need for further molecular subclassification of these gastric cancer subtypes in order to elucidate mechanisms of primary resistance to immunotherapy.
9. Conclusions
In conclusion, immunotherapy has been incorporated in the clinical management of advanced GC based on recent positive phase III clinical trial results. Most of the benefit is seen in PD-L1 CPS positive, MSI-H/MMR-D, EBV-positive, and possibly TMB-high tumours. Although these biomarkers have been associated with durable responses to immunotherapy, there is still a considerable proportion of patients who fail to respond, while ultimately cancer cells develop immune-evasion strategies and develop resistance. Further clinical combined with translational research is eagerly needed to optimise immunotherapy efficacy and overcome emerging PD-1/PD-L1 resistance in order to improve GC patients’ outcomes.