 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | LAURA CONTI | + 2271 word(s) | 2271 | 2020-12-16 07:59:32 | | | |
| 2 | Nicole Yin | Meta information modification | 2271 | 2020-12-18 09:42:09 | | |
Video Upload Options
TLRs are one of four major families of pattern recognition receptor (PRRs), which include also NOD-like receptors (NLRs), RIG-like receptors (RLRs), C-type lectin receptors (CLRs), and represent the cornerstone of the innate immune response. TLR2, together with TLR1, TLR3, TLR4, and TLR5, was first identified and characterized in 1998. TLR2 is the only TLR that forms functional heterodimers with more than two other types of TLRs, forming dimers with TLR1, TLR6, and in some cases with TLR4. TLR2 recognizes molecules frequently associated with pathogens, the so-called pathogen-associated molecular patterns (PAMPs), leading to activation of downstream signal transduction pathways, which result in the production of inflammatory cytokines, type I interferons (IFNs), and other mediators necessary for the development of effective immune responses. Moreover, TLR2 is involved in the recognition of damage-associated molecular patterns (DAMPs), released by damaged tissues.
1. Introduction
TLR2 interacts also with different non-TLR molecules, allowing the recognition of a great variety of PAMPs from all microbial phyla including viruses, fungi, bacteria, and parasites. In particular, TLR2 can sense highly conserved lipoproteins expressed on the outer membranes of Gram-positive bacteria and, in association with its co-receptor CD14, membrane antigens of some Gram-negative bacteria, such as lipopolysaccharide (LPS). Independently from the dimers formed, the downstream signaling cascade proceeds through the MyD88-dependent pathway, leading to cytokine production[1].
TLR2 is expressed by immune cells, endothelial cells, and epithelial cells. This wide expression is consistent with the widespread range of roles and functions of TLR2[2]. A beneficial role for TLR2 signaling is described in the maintenance of mucosal homeostasis and defense against some pathogens, whereas TLR2 signaling stimulated by other pathogens or after endogenous activation is correlated with a more severe phenotype in infectious and inflammatory diseases. In addition, there is a clear role for TLRs in microbe–host and host–microbe interactions, thus mediating the cross-talk with tissue microbiota[3]. Moreover, TLR2 activation by DAMPs released from damaged tissues mediates a kind of inflammatory response. For example, TLR2 can be activated by extracellular matrix components, cytoplasmic proteins as S100 or HSPs, and nuclear structural proteins such as histones and HMGB1[4]. Thus, TLR2’s ability to interact with several self or non-self molecules confers it a key role not only in anti-pathogens response but also in the maintenance of the homeostatic conditions. Several types of cells in the body can dialogue through TLR2 sensing and activation. The correct interplay between tissue cells, immune cells, and microbiota is a necessary condition for a healthy organism. However, this equilibrium can be lost, for example as a consequence of aggressive therapies (i.e., antibiotics or chemotherapy), microbiota alterations, or other disorders, such as cancer[3]. In that case, TLR2 may have a dual role and be a central element in the anti-tumor immune response or in the pro-tumorigenic processes. A deeper comprehension of these two mechanisms is essential for the development of new approaches for the design of anti-cancer therapies.
2. The Role of TLR2 in Anti-Tumor Immune Response
Lymphoid and myeloid cells present in the TME can have either an immune suppressive or immune stimulatory function, being therefore important regulators of cancer progression, survival, as well as therapy resistance[5]. The presence of dying cells in the TME causes the release of DAMPs, which in turn activate myeloid cells thanks to their PRRs[6]. For instance, HMGB1, a nuclear protein that upon cell damage or death can be released in the TME, is able to stimulate several PRRs signaling, including TLR2[7]. DCs can be activated by DAMPs together with other stimuli, such as TNF-α and type I IFNs. Once stimulated, DCs enhance their antigen-presenting activity by increasing the expression of MHC class II and costimulatory molecules, resulting in the activation of an adaptive immune response[8]. To study whether TLR2 activation may promote anti-cancer immune responses, exogenous TLR2 ligands were administered in several murine cancer models. In lung cancer models, the administration of synthetic or bacterial-derived TLR2 ligands induces the differentiation of monocytic myeloid derived suppressor cells (M-MDSC) toward the anti-tumoral M1 phenotype, with the production of IFN-γ, nitric oxide (NO), and pro-inflammatory cytokines such as TNF-α, IL-12p40, and IL-12p70, suggesting a correlation between TLR2 activation and a better anti-tumor response[9][10]. The efficacy of TLR2 stimulation on tumor-infiltrated leukocytes was demonstrated also in a melanoma model. Indeed, it was reported that the administration of the synthetic compound diprovocim activates the TLR1/2 signaling and synergizes with ovalbumin (OVA) immunization and anti-programmed death-ligand (PD-L)1 treatment in mice engrafted with B16-OVA, promoting leukocyte infiltration in tumor as well as an increased systemic humoral and cytotoxic response[11]. Contrasting results were observed in a study on fibrosarcoma in vivo. Mice immunized with tumor-associated antigens (TAAs) together with the synthetic lipoprotein FSL-1, a TLR2/6 ligand, showed a significant decrease of tumor growth due to the induction of T helper type 2 (Th2) cells. This combined treatment produced an increase in tumor-specific CTLs and a humoral response able to mediate antibody-dependent cell-mediated cytotoxicity (ADCC), tumor lysis by complement activation, and a reduction of Treg cell frequency. However, the treatment with FSL-1 alone increased the number of Tregs and enhanced tumor growth. This effect was reverted by the combined administration of anti-CD25 antibodies, suggesting the involvement of Tregs in the FSL-1 pro-tumoral effect. None of these effects was observed in TLR2-/- mice, confirming that both pro- and anti-tumor effects are TLR2-dependent[12]. TLR2 seems to play a role also in NK cell-mediated anti-tumor immunity. Indeed, in lung cancer-bearing mice, TLR2 stimulation by treatment with the natural polysaccharide SEP induces NK cell activation, proliferation, cancer cell-directed cytotoxicity, and the release of IL-2 and IFN-γ[13]. Furthermore, TLR2 stimulation with the natural polysaccharide krestin (PSK) activates human NK cells, inducing IFN-γ secretion and the lysis of K562 cells. In addition, PSK also enhances the efficacy of trastuzumab on SKBR3 cells as well as on Her2/Neu transgenic mice by potentiating the ADCC by NK cells[14].
However, although the role of TLR2 in tumors has been widely studied, it remains ambiguous. Indeed, there is a huge variation on the results obtained by different groups, depending on several factors, such as the type of tumor and the model used, with reports suggesting that TLR2 signaling can activate an anti-tumor immune response, while others highlight its immune suppressive function, as described in the next sections.
3. The Role of TLR2 in Immunosuppression
As discussed in the previous paragraph, TLR2 plays an important role in the activation of innate immune cells and may promote anti-tumor responses in virtue of its ability to activate antigen presenting cells and, consequently, tumor-specific T lymphocytes. However, TLR2 is also expressed on immune cells, such as Tregs, MDSCs, macrophages, and neutrophils, which can contribute to tumor growth and metastatic dissemination by stimulating the generation of an immunosuppressive microenvironment and of the metastatic niche[15]. Of note, TLR2 activation promotes their immunosuppressive function, thus favoring tumor growth[16] (Figure 1).
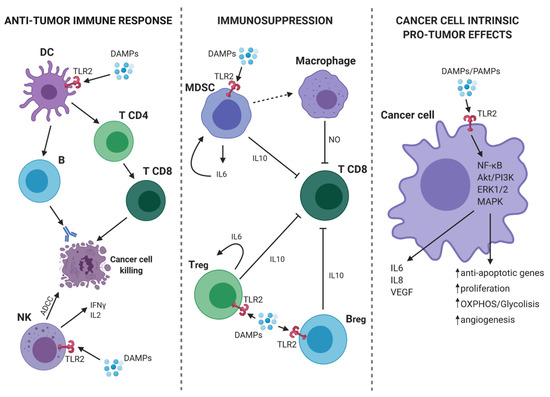
Figure 1. TLR2-mediated pro- and anti-tumor mechanisms. (On the left) In tumor microenvironment (TME), the presence of DAMPs activates dendritic cells (DCs) through TLR2. Consequently, DCs present the antigen and initiate a specific anti-tumor T cell response. At the same time, TLR2 expressed on Natural Killer (NK) cells mediates their activation and the induction of antibody-dependent cell-mediated cytotoxicity (ADCC) against cancer cells as well as the release of IFN-γ and interleukin (IL)-2. (On the center) TLR2 activation on myeloid-derived suppressor cells (MDSCs), Bregs and Tregs leads to an immunosuppressive effect. Indeed, MDSCs can induce macrophages to release nitric oxide (NO) that suppresses the activity of T CD8+ lymphocytes. Moreover, activated MDSCs, Bregs and T regulatory cells (Tregs), release IL-6 and IL-10 with the consequent synergic inhibitory effect on T CD8+ cells. (On the right) TLR2 expressed on cancer cells can bind DAMPs/PAMPs and activate intracellular signaling, such as nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), Akt/PI3K, extracellular signal-regulated kinase (ERK)1/2, and MAPK. The consequence is a pro-tumor effect due to the release of IL-6, IL-8, VEGF, and the promotion of proliferation, cell metabolism, angiogenesis, and protection from apoptosis. Continous and dashed arrows indicate activation or secretion. T-arrows indicate cellular inhibition. Created with BioRender.com.
Since the characterization of the role exerted by TLR2 on immunosuppressive cell populations may provide therapeutic tools for the treatment of both autoimmune and neoplastic diseases, many researchers have investigated the effects of TLR2 signaling in MDSCs. These important studies demonstrated that TLR2 activation drives the differentiation of MDSCs from bone marrow progenitor cells and promotes their survival and proliferation by activating NF-κB and MAPK and inducing the production of IL-6 and the consequent activation of the signal transducer and activator of transcription (STAT3). Therefore, TLR2 activation increases MDSC accumulation in the tumor microenvironment and in lymphoid organs[17]. The same group of researchers subsequently demonstrated that TLR2 activation may promote the immunosuppressive activity of monocytic-MDSCs, inducing their differentiation into tumor-promoting macrophages. The underlying mechanism is very intriguing, since it involves the collaboration of tumor-infiltrating CD8+ T cells, which are the final victims of MDSC function. Indeed, MDSC-derived macrophages present antigenic peptides to CD8+ T cells, inducing their transient activation and the production of IFN-γ. Then, this cytokine leads to inducible nitric oxide synthase (iNOS) expression in macrophages, and iNOS-generated NO inhibits T cell proliferation induced by DCs[18]. Furthermore, TLR2 activation stimulates MDSCs to produce IL-10, which induces macrophage polarization toward the M2 tumor-promoting phenotype, while MDSC-produced IL-6 promotes tumor metastatic growth[19][20].
The immunosuppressive effect of TLR2 activation is not limited to MDSCs but takes place also in DCs. In fact, in the TME, cancer cells promote TLR2 activation in DCs, which induces the secretion of elevated levels of cytokines including IL-6 and IL-10 and the overexpression of their receptors. Thus, these cytokines act in an autocrine manner to activate STAT3 in DCs and reduce the expression of IL-12, MHC class II, CD40, and CD86. These dysfunctional DCs lose the ability to activate anti-cancer cytotoxic T cells while promoting the function of Treg cells[21]. These results suggested that TLR2 could indirectly influence the activity of Treg through a DC-mediated mechanism but did not exclude the presence of Treg cell-intrinsic effects. This gap was filled by a very interesting paper by Sutmuller et al. that, analyzing the direct effects induced by TLR2 agonists on Treg and T helper cells in vitro, showed that TLR2 activation in Treg cells induces their proliferation and may directly stimulate their immunosuppressive activity[22]. Physiologically, this mechanism is activated by commensal bacterial components of the microbiota that mediate the TLR2-dependent conversion of CD4+ T cells into forkhead box P3 (Foxp3)+ Treg cells that produce IL-10, with the aim of inducing mucosal tolerance[23]. These studies demonstrated the importance of TLR2 in immune system homeostasis and in the balance between tolerance and immunity, but no indication of the consequences that this immunosuppressive mechanism may have on tumorigenesis and cancer progression were provided. Therefore, several research groups addressed the question whether, in the tumor context, TLR2 can exert detrimental effects, promoting the suppression of effector T cell function. A seminal paper published in 2019 by the group of Prof. Wang demonstrated that secretory autophagosomes containing the TLR2 ligand HSP90α, which is released by cancer cells as a result of an alternative non-degradative autophagy mechanism used for the unconventional secretion of small molecules, stimulate IL-6 production in CD4+ T lymphocytes through the TLR2–MyD88–NF-κB axis. IL-6 promotes the production of IL-10 and IL-21 in an autocrine manner, conferring regulatory and immunosuppressive activity to T cells [40]. These data are very important, since they clearly demonstrate that cancer cells can induce TLR2 activation to interfere with host anti-tumor immunity. However, the role of TLR2 on Treg function is controversial, since other researchers demonstrated that TLR2 activation on Tregs transiently inhibits their immunosuppressive activity[22]. In our opinion, these data are not in contrast but reflect the multifaceted nature of TLR2, whose effects might be context-dependent.
In addition to myeloid and T cells, TLR2 is likewise expressed on B lymphocytes and induces their differentiation toward the T cell Ig and mucin domain (TIM)-1 expressing regulatory B cells (Bregs)[24] through the activation of the MAPK pathway. These Bregs produce high amounts of IL-10 and play a key role in the inhibition of effector T cell function and proliferation. A high infiltration of TIM-1+ B cells correlates with advanced disease stage and poor overall survival in patients with hepatocellular carcinoma[25].
The activation of TLR2 on tumor infiltrating myeloid or Treg cells is induced directly by cancer cells, which are able to secrete TLR2 endogenous ligands, such as the chondroitin sulfate proteoglycan versican, HMGB1 (which will be discussed in the following paragraphs) and Wnt5a[19][26]. This intercellular communication does not only take place in the TME, as tumor cells can influence immune cells at distant sites. Indeed, tumor cells secrete exosomes and secretory autophagosomes carrying endogenous TLR2 ligands (HMGB1, HSP72, HSP90, palmitoylated proteins and others)[24], which reach distant organs and, by activating the TLR2–IL-6–STAT3 signaling pathway in myeloid cells, promote the establishment of the pre-metastatic niche and contribute to the “seed and soil” mechanism responsible for metastasis development[27]. The pro-metastatic activity of TLR2 is confirmed by the fact that TLR2 knockout mice develop fewer lung, liver, and adrenal metastases following the subcutaneous implantation of cancer cells as compared to their wild-type counterpart[19][26].
Thus, we can conclude that TLR2 activation on immunosuppressive cells plays an important role for the maintenance of immune system homeostasis in physiological conditions but can exert detrimental effects when this happens in the context of carcinogenesis. Further studies are needed to verify whether the PAMP/DAMP/TLR2 axis may represent a safe and effective target for cancer treatment.
References
- van Bergenhenegouwen, J.; Plantinga, T.S.; Joosten, L.A.; Netea, M.G.; Folkerts, G.; Kraneveld, A.D.; Garssen, J.; Vos, A.P.; TLR2 & Co: A critical analysis of the complex interactions between TLR2 and coreceptors. J. Leukoc. Biol. 2013, 94, 885–902.
- Laura Oliveira-Nascimento; Paola Massari; Lee M Wetzler; The Role of TLR2 in Infection and Immunity. Frontiers in Immunology 2012, 3, 79, 10.3389/fimmu.2012.00079.
- Danping Zheng; Timur Liwinski; Eran Elinav; Interaction between microbiota and immunity in health and disease. Cell Research 2020, 30, 492-506, 10.1038/s41422-020-0332-7.
- Jong Seong Roh; Dong Hyun Sohn; Damage-Associated Molecular Patterns in Inflammatory Diseases. Immune Network 2018, 18, e27, 10.4110/in.2018.18.e27.
- Laura Conti; Roberto Ruiu; Giuseppina Barutello; Marco Macagno; Silvio Bandini; Federica Cavallo; Stefania Lanzardo; Microenvironment, Oncoantigens, and Antitumor Vaccination: Lessons Learned from BALB-neuT Mice. BioMed Research International 2014, 2014, 1-16, 10.1155/2014/534969.
- Hajime Kono; Kenneth L. Rock; How dying cells alert the immune system to danger. Nature Reviews Immunology 2008, 8, 279-289, 10.1038/nri2215.
- Rui Kang; Qiuhong Zhang; Herbert J. Zeh; Michael T. Lotze; Daolin Tang; HMGB1 in cancer: good, bad, or both?. Clinical Cancer Research 2013, 19, 4046-4057, 10.1158/1078-0432.CCR-13-0495.
- Olivier Joffre; Martijn A. Nolte; Roman Spörri; Caetano Reis E Sousa; Inflammatory signals in dendritic cell activation and the induction of adaptive immunity. Immunological Reviews 2009, 227, 234-247, 10.1111/j.1600-065x.2008.00718.x.
- Deng, Y.; Yang, J.; Qian, J.; Liu, R.; Huang, E.; Wang, Y.; Luo, F.; Chu, Y. TLR1/TLR2 signaling blocks the suppression of monocytic myeloid-derived suppressor cell by promoting its differentiation into M1-type macrophage. Mol. Immunol. 2019, 112, 266–273.
- Muller, E.; Christopoulos, P.F.; Halder, S.; Lunde, A.; Beraki, K.; Speth, M.; Oynebraten, I.; Corthay, A. Toll-Like Receptor Ligands and Interferon-gamma Synergize for Induction of Antitumor M1 Macrophages. Front. Immunol. 2017, 8, 1383.
- Ying Wang; Lijing Su; Matthew D. Morin; Brian T. Jones; Yuto Mifune; Hexin Shi; Kuan-Wen Wang; XiaoMing Zhan; Aijie Liu; Jianhui Wang; et al.Xiaohong LiMiao TangSara LudwigSara HildebrandKejin ZhouDaniel J. SiegwartEva Marie Y. MorescoHong ZhangDale L. BogerBruce Beutler Adjuvant effect of the novel TLR1/TLR2 agonist Diprovocim synergizes with anti–PD-L1 to eliminate melanoma in mice. Proceedings of the National Academy of Sciences 2018, 115, E8698-E8706, 10.1073/pnas.1809232115.
- Kazuto Kiura; Akira Hasebe; Ayumi Saeki; Taku Segawa; Futoshi Okada; Haque Mohammad Shamsul; Makoto Ohtani; Takeshi Into; Nobuo Inoue; Minoru Wakita; et al.Ken‐Ichiro Shibata In vivo anti- and pro-tumour activities of the TLR2 ligand FSL-1. Immunobiology 2011, 216, 891-900, 10.1016/j.imbio.2011.02.006.
- Mengyun Ke; Hui Wang; Min Zhang; Yuwei Tian; Yizhou Wang; Bing Li; Jie Yu; Jie Dou; Tao Xi; Changlin Zhou; et al. The anti-lung cancer activity of SEP is mediated by the activation and cytotoxicity of NK cells via TLR2/4 in vivo. Biochemical Pharmacology 2014, 89, 119-130, 10.1016/j.bcp.2014.02.024.
- Hailing Lu; Y. Yang; Ekram Gad; Carol Inatsuka; Cynthia A. Wenner; Mary L. Disis; Leanna J. Standish; TLR2 Agonist PSK Activates Human NK Cells and Enhances the Antitumor Effect of HER2-Targeted Monoclonal Antibody Therapy. Clinical Cancer Research 2011, 17, 6742-6753, 10.1158/1078-0432.CCR-11-1142.
- Kanako Shimizu; Tomonori Iyoda; Masahiro Okada; Satoru Yamasaki; Shin-Ichiro Fujii; Immune suppression and reversal of the suppressive tumor microenvironment. International Immunology 2018, 30, 445-455, 10.1093/intimm/dxy042.
- Sabina Kaczanowska; Ann Mary Joseph; Eduardo Davila; TLR agonists: our best frenemy in cancer immunotherapy. Journal of Leukocyte Biology 2013, 93, 847-863, 10.1189/jlb.1012501.
- Akira Maruyama; Hiroaki Shime; Yohei Takeda; Masahiro Azuma; Misako Matsumoto; Tsukasa Seya; Pam2 lipopeptides systemically increase myeloid-derived suppressor cells through TLR2 signaling. Biochemical and Biophysical Research Communications 2015, 457, 445-450, 10.1016/j.bbrc.2015.01.011.
- Hiroaki Shime; Akira Maruyama; Sumito Yoshida; Yohei Takeda; Misako Matsumoto; Tsukasa Seya; Toll-like receptor 2 ligand and interferon-γ suppress anti-tumor T cell responses by enhancing the immunosuppressive activity of monocytic myeloid-derived suppressor cells. OncoImmunology 2017, 7, e1373231, 10.1080/2162402X.2017.1373231.
- Kim, S.; Takahashi, H.; Lin, W.W.; Descargues, P.; Grivennikov, S.; Kim, Y.; Luo, J.L.; Karin, M. Carcinoma-produced factors activate myeloid cells through TLR2 to stimulate metastasis. Nature 2009, 457, 102–106.
- Sinha, P.; Clements, V.K.; Bunt, S.K.; Albelda, S.M.; Ostrand-Rosenberg, S. Cross-talk between myeloid-derived suppressor cells and macrophages subverts tumor immunity toward a type 2 response. J. Immunol. 2007, 179, 977–983.
- Michael Tang; Jun Diao; Hongtao Gu; Ismat Khatri; Jun Zhao; Mark S. Cattral; Toll-like Receptor 2 Activation Promotes Tumor Dendritic Cell Dysfunction by Regulating IL-6 and IL-10 Receptor Signaling. Cell Reports 2015, 13, 2851-2864, 10.1016/j.celrep.2015.11.053.
- Roger P.M. Sutmuller; Martijn H.M.G.M. Den Brok; Matthijs Kramer; Erik J. Bennink; Liza W.J. Toonen; Bart-Jan Kullberg; Leo A. Joosten; Shizuo Akira; Mihai G. Netea; G. J. Adema; et al. Toll-like receptor 2 controls expansion and function of regulatory T cells. Journal of Clinical Investigation 2006, 116, 485-494, 10.1172/jci25439.
- June L. Round; Sarkis K. Mazmanian; Inducible Foxp3+ regulatory T-cell development by a commensal bacterium of the intestinal microbiota. Proceedings of the National Academy of Sciences 2010, 107, 12204-12209, 10.1073/pnas.0909122107.
- Yongqiang Chen; Peng-Cheng Li; Ning Pan; Rong Gao; Zhi-Fa Wen; Tianyu Zhang; Fang Huang; Fang-Yuan Wu; Xi-Long Ou; Jinping Zhang; et al.Xuejun ZhuHong-Ming HuKang ChenYun-Lang CaiLi-Xin Wang Tumor-released autophagosomes induces CD4+ T cell-mediated immunosuppression via a TLR2–IL-6 cascade. Journal for ImmunoTherapy of Cancer 2019, 7, 178, 10.1186/s40425-019-0646-5.
- Linsen Ye; Qi Zhang; Yusheng Cheng; Xiaolong Chen; Guoying Wang; Mengchen Shi; Tong Zhang; Yingjiao Cao; Hang Pan; Liting Zhang; et al.Genshu WangYinan DengYang YangGuihua Chen Tumor-derived exosomal HMGB1 fosters hepatocellular carcinoma immune evasion by promoting TIM-1+ regulatory B cell expansion. Journal for ImmunoTherapy of Cancer 2018, 6, 145, 10.1186/s40425-018-0451-6.
- Meliha Mehmeti; Caroline Bergenfelz; Eva Källberg; Camilla Rydberg Millrud; Per Björk; Fredrik Ivars; Bengt Johansson-Lindbom; Sven Kjellström; Ingemar André; Karin Leandersson; et al. Wnt5a is a TLR2/4-ligand that induces tolerance in human myeloid cells. Communications Biology 2019, 2, 176, 10.1038/s42003-019-0432-4.
- Amy Chow; Weiying Zhou; Liang Liu; Miranda Y. Fong; Jackson Champer; Desiree Van Haute; Andrew R. Chin; Xiubao Ren; Bogdan Gabriel Gugiu; Zhipeng Meng; et al.Wendong HuangVu NgoMarcin KortylewskiShizhen Emily Wang Macrophage immunomodulation by breast cancer-derived exosomes requires Toll-like receptor 2-mediated activation of NF-κB. Scientific Reports 2014, 4, srep05750, 10.1038/srep05750.

