 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Kathy Nga-Chu LUI | -- | 2067 | 2022-11-08 08:37:09 | | | |
| 2 | Beatrix Zheng | + 2 word(s) | 2069 | 2022-11-09 02:51:19 | | |
Video Upload Options
Hirschsprung disease (HSCR) is a complex congenital disorder caused by defects in the development of the enteric nervous system (ENS). It is attributed to failures of the enteric neural crest stem cells (ENCCs) to proliferate, differentiate and/or migrate, leading to the absence of enteric neurons in the distal colon, resulting in colonic motility dysfunction. Due to the oligogenic nature of the disease, some HSCR conditions could not be phenocopied in animal models. Building the patient-based disease model using human induced pluripotent stem cells (hPSC) has opened up a new opportunity to untangle the unknowns of the disease. The expanding armamentarium of hPSC-based therapies provides needed new tools for developing cell-replacement therapy for HSCR.
1. Introduction
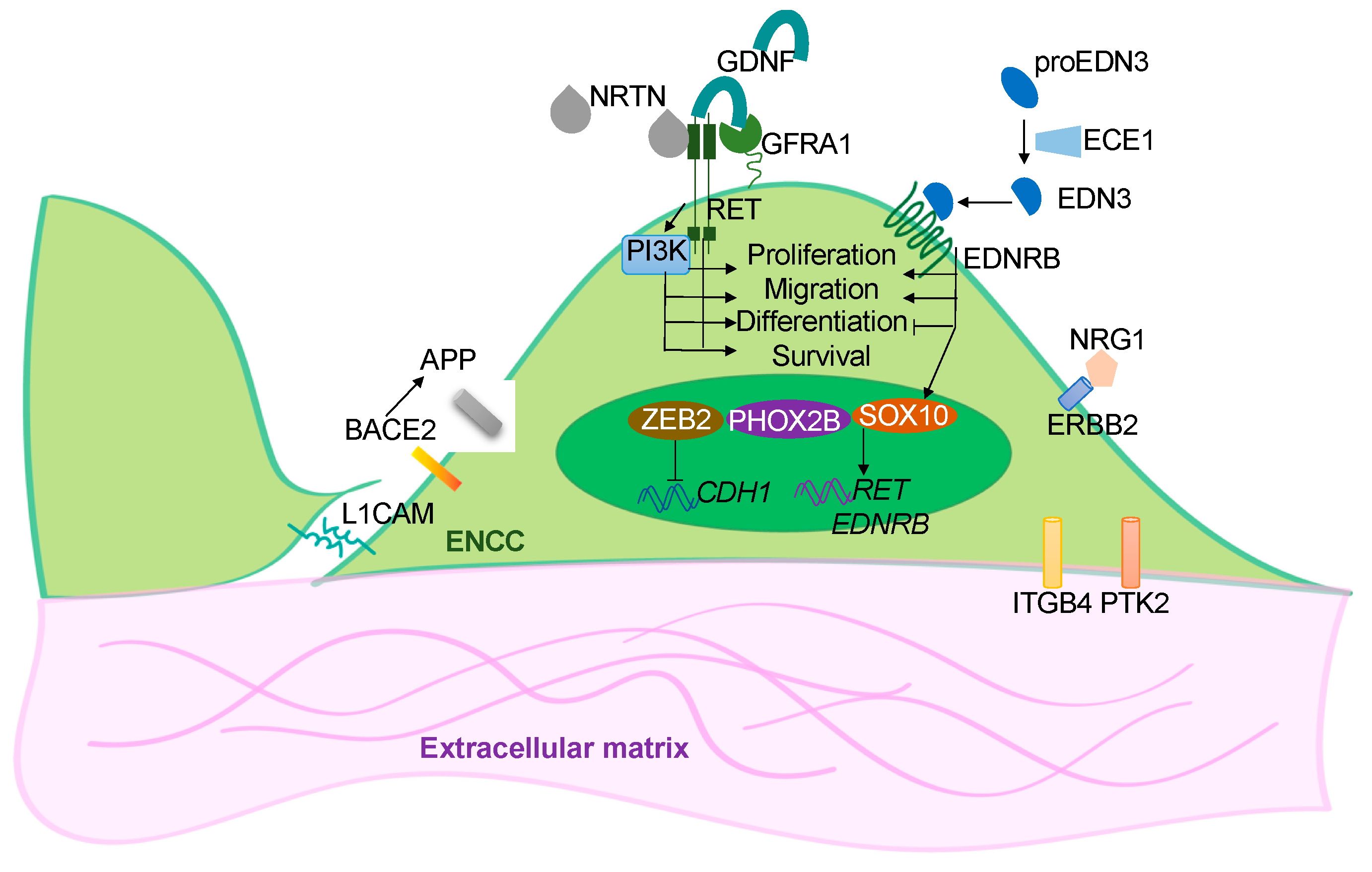
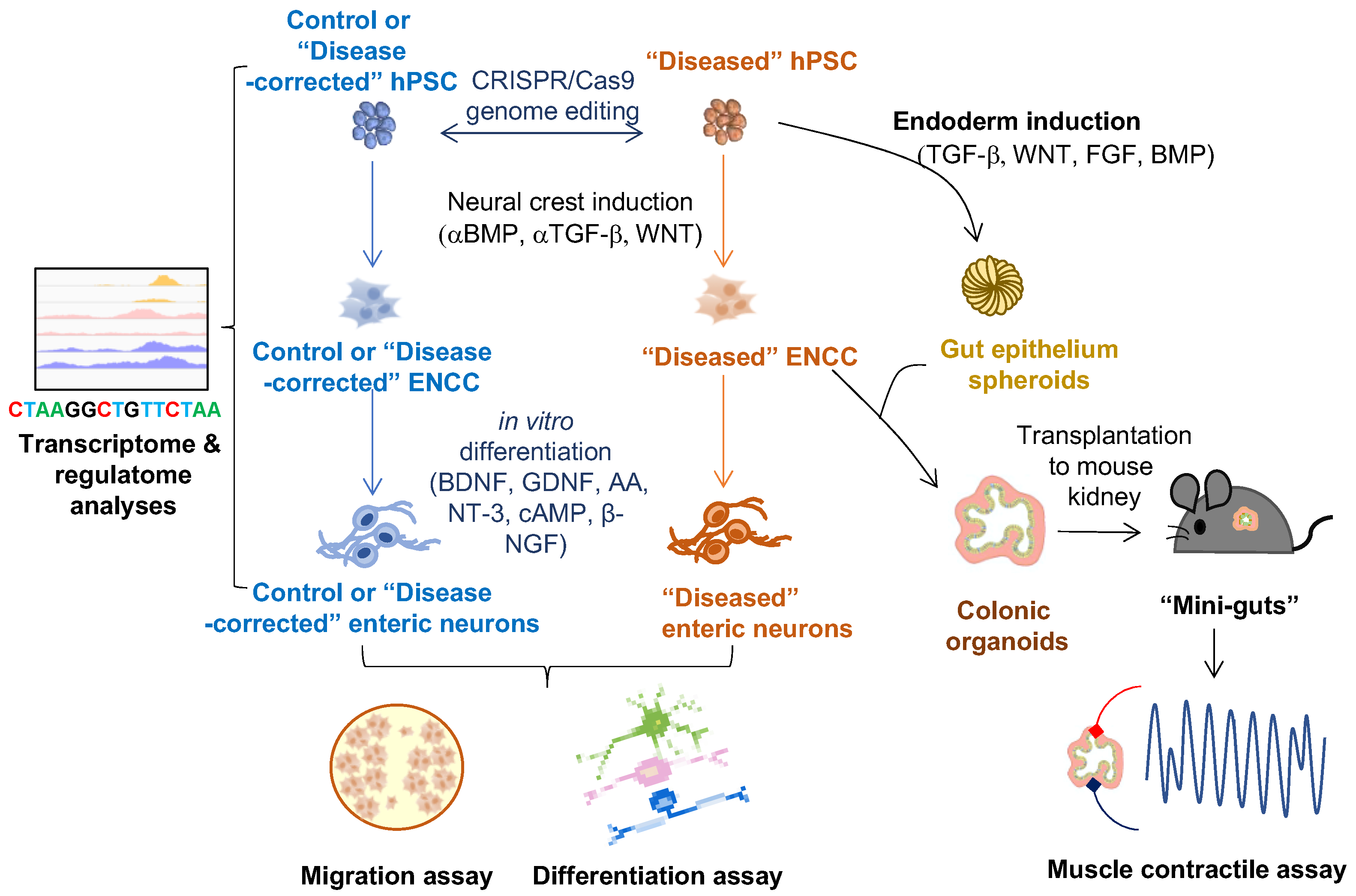
2. hPSC-Based Disease Models of HSCR
2.1. In Vitro 2-D ENS Model
2.2. 3-D Human Colonic Organoids
References
- Tilghman, J.M.; Ling, A.Y.; Turner, T.N.; Sosa, M.X.; Krumm, N.; Chatterjee, S.; Kapoor, A.; Coe, B.P.; Nguyen, K.H.; Gupta, N.; et al. Molecular Genetic Anatomy and Risk Profile of Hirschsprung’s Disease. N. Engl. J. Med. 2019, 380, 1421–1432.
- Sergi, C. Hirschsprung’s disease: Historical notes and pathological diagnosis on the occasion of the 100(th) anniversary of Dr. Harald Hirschsprung’s death. World J. Clin. Pediatr. 2015, 4, 120–125.
- Heuckeroth, R.O. Hirschsprung disease-integrating basic science and clinical medicine to improve outcomes. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 152–167.
- Goldstein, A.M.; Thapar, N.; Karunaratne, T.B.; De Giorgio, R. Clinical aspects of neurointestinal disease: Pathophysiology, diagnosis, and treatment. Dev. Biol. 2016, 417, 217–228.
- Badner, J.A.; Sieber, W.K.; Garver, K.L.; Chakravarti, A. A Genetic-Study of Hirschsprung Disease. Am. J. Hum. Genet. 1990, 46, 568–580.
- Tang, C.S.M.; Li, P.; Lai, F.P.L.; Fu, A.X.; Lau, S.T.; So, M.T.; Lui, K.N.C.; Li, Z.X.; Zhuang, X.H.; Yu, M.; et al. Identification of Genes Associated With Hirschsprung Disease, Based on Whole-Genome Sequence Analysis, and Potential Effects on Enteric Nervous System Development. Gastroenterology 2018, 155, 1908−+.
- Chow, K.H.-M.; Tam, P.K.-H.; Ngan, E.S.-W. Neural Crest and Hirschsprung’s Disease. In Stem Cells and Human Diseases; Srivastava, R.K., Shankar, S., Eds.; Springer: Dordrecht, The Netherlands, 2012; pp. 353–386.
- Obermayr, F.; Hotta, R.; Enomoto, H.; Young, H.M. Development and developmental disorders of the enteric nervous system. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 43–57.
- Heanue, T.A.; Pachnis, V. Enteric nervous system development and Hirschsprung’s disease: Advances in genetic and stem cell studies. Nat. Rev. Neurosci. 2007, 8, 466–479.
- Lake, J.I.; Heuckeroth, R.O. Enteric nervous system development: Migration, differentiation, and disease. Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 305, G1–G24.
- Lee, G.; Chambers, S.M.; Tomishima, M.J.; Studer, L. Derivation of neural crest cells from human pluripotent stem cells. Nat. Protoc. 2010, 5, 688–701.
- Barber, K.; Studer, L.; Fattahi, F. Derivation of enteric neuron lineages from human pluripotent stem cells. Nat. Protoc. 2019, 14, 1261–1279.
- Fattahi, F.; Steinbeck, J.A.; Kriks, S.; Tchieu, J.; Zimmer, B.; Kishinevsky, S.; Zeltner, N.; Mica, Y.; El-Nachef, W.; Zhao, H.; et al. Deriving human ENS lineages for cell therapy and drug discovery in Hirschsprung disease. Nature 2016, 531, 105–109.
- Kwart, D.; Paquet, D.; Teo, S.; Tessier-Lavigne, M. Precise and efficient scarless genome editing in stem cells using CORRECT. Nat. Protoc. 2017, 12, 329–354.
- Mali, P.; Yang, L.; Esvelt, K.M.; Aach, J.; Guell, M.; DiCarlo, J.E.; Norville, J.E.; Church, G.M. RNA-guided human genome engineering via Cas9. Science 2013, 339, 823–826.
- Paquet, D.; Kwart, D.; Chen, A.; Sproul, A.; Jacob, S.; Teo, S.; Olsen, K.M.; Gregg, A.; Noggle, S.; Tessier-Lavigne, M. Efficient introduction of specific homozygous and heterozygous mutations using CRISPR/Cas9. Nature 2016, 533, 125–129.
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308.
- Fu, A.X.; Lui, K.N.; Tang, C.S.; Ng, R.K.; Lai, F.P.; Lau, S.T.; Li, Z.; Garcia-Barcelo, M.M.; Sham, P.C.; Tam, P.K.; et al. Whole-genome analysis of noncoding genetic variations identifies multiscale regulatory element perturbations associated with Hirschsprung disease. Genome Res. 2020, 30, 1618–1632.
- Lai, F.P.; Lau, S.T.; Wong, J.K.; Gui, H.; Wang, R.X.; Zhou, T.; Lai, W.H.; Tse, H.F.; Tam, P.K.; Garcia-Barcelo, M.M.; et al. Correction of Hirschsprung-Associated Mutations in Human Induced Pluripotent Stem Cells Via Clustered Regularly Interspaced Short Palindromic Repeats/Cas9, Restores Neural Crest Cell Function. Gastroenterology 2017, 153, 139–153.e8.
- McCracken, K.W.; Cata, E.M.; Crawford, C.M.; Sinagoga, K.L.; Schumacher, M.; Rockich, B.E.; Tsai, Y.H.; Mayhew, C.N.; Spence, J.R.; Zavros, Y.; et al. Modelling human development and disease in pluripotent stem-cell-derived gastric organoids. Nature 2014, 516, 400–404.
- Spence, J.R.; Mayhew, C.N.; Rankin, S.A.; Kuhar, M.F.; Vallance, J.E.; Tolle, K.; Hoskins, E.E.; Kalinichenko, V.V.; Wells, S.I.; Zorn, A.M.; et al. Directed differentiation of human pluripotent stem cells into intestinal tissue in vitro. Nature 2011, 470, 105–109.
- Workman, M.J.; Mahe, M.M.; Trisno, S.; Poling, H.M.; Watson, C.L.; Sundaram, N.; Chang, C.F.; Schiesser, J.; Aubert, P.; Stanley, E.G.; et al. Engineered human pluripotent-stem-cell-derived intestinal tissues with a functional enteric nervous system. Nat. Med. 2017, 23, 49–59.
- Schlieve, C.R.; Fowler, K.L.; Thornton, M.; Huang, S.; Hajjali, I.; Hou, X.; Grubbs, B.; Spence, J.R.; Grikscheit, T.C. Neural Crest Cell Implantation Restores Enteric Nervous System Function and Alters the Gastrointestinal Transcriptome in Human Tissue-Engineered Small Intestine. Stem Cell Rep. 2017, 9, 883–896.
- Lau, S.T.; Li, Z.; Lai, F.P.L.; Lui, K.N.C.; Li, P.; Múnera, J.O.; Pan, G.; Mahe, M.M.; Hui, C.C.; Wells, J.M.; et al. Activation of Hedgehog Signaling Promotes Development of Mouse and Human Enteric Neural Crest Cells, Based on Single-Cell Transcriptome Analyses. Gastroenterology 2019, 157, 1556–1571.e5.
- Múnera, J.O.; Sundaram, N.; Rankin, S.A.; Hill, D.; Watson, C.; Mahe, M.; Vallance, J.E.; Shroyer, N.F.; Sinagoga, K.L.; Zarzoso-Lacoste, A.; et al. Differentiation of Human Pluripotent Stem Cells into Colonic Organoids via Transient Activation of BMP Signaling. Cell Stem Cell 2017, 21, 51–64.e6.
- Crespo, M.; Vilar, E.; Tsai, S.Y.; Chang, K.; Amin, S.; Srinivasan, T.; Zhang, T.; Pipalia, N.H.; Chen, H.J.; Witherspoon, M.; et al. Colonic organoids derived from human induced pluripotent stem cells for modeling colorectal cancer and drug testing. Nat. Med. 2017, 23, 878–884.
- Eicher, A.K.; Kechele, D.O.; Sundaram, N.; Berns, H.M.; Poling, H.M.; Haines, L.E.; Sanchez, J.G.; Kishimoto, K.; Krishnamurthy, M.; Han, L.; et al. Functional human gastrointestinal organoids can be engineered from three primary germ layers derived separately from pluripotent stem cells. Cell Stem Cell 2022, 29, 36–51.

