 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Esther García-Domínguez | -- | 2931 | 2022-10-07 18:45:09 | | | |
| 2 | Lindsay Dong | -3 word(s) | 2928 | 2022-11-02 10:12:46 | | |
Video Upload Options
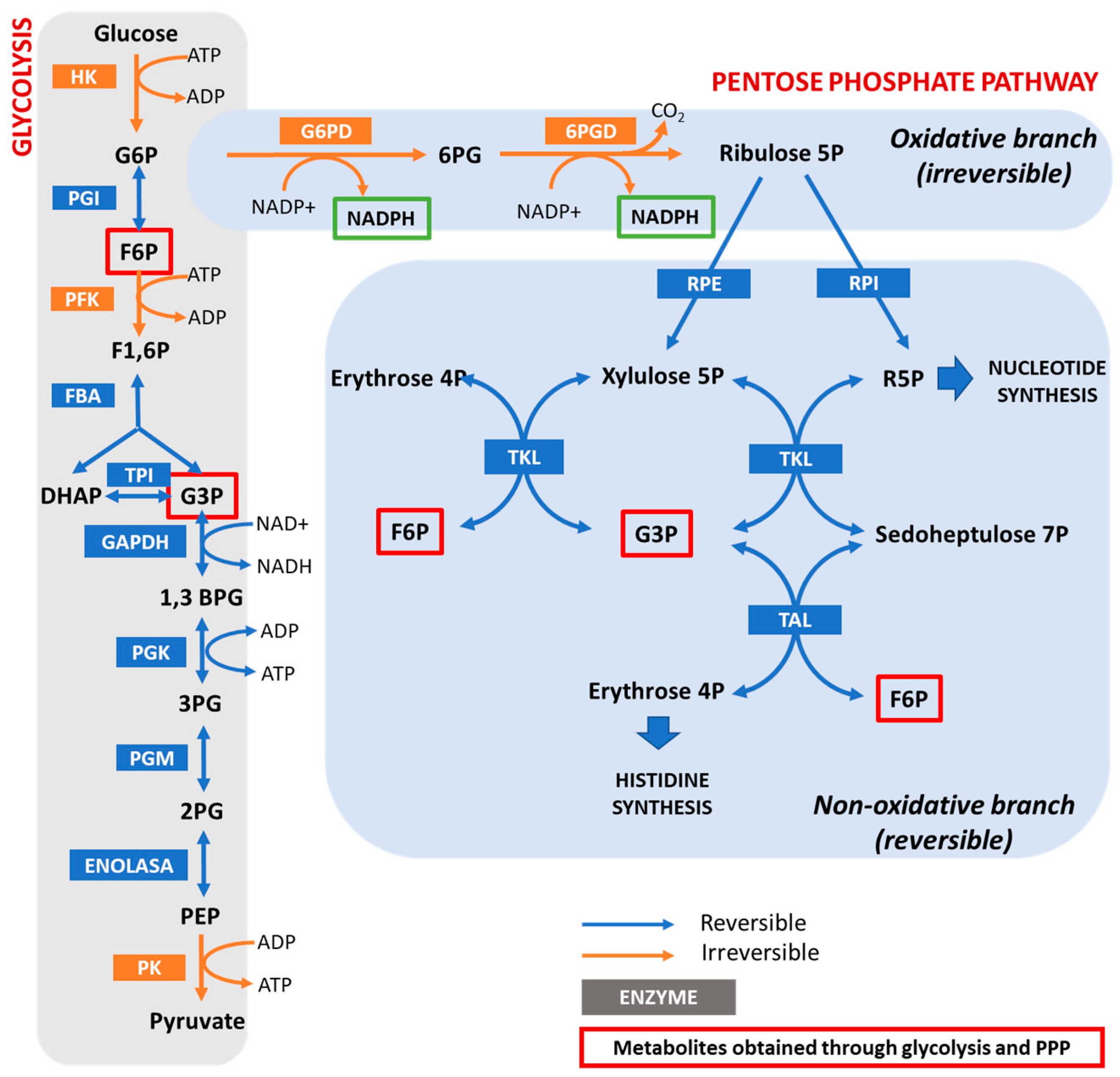
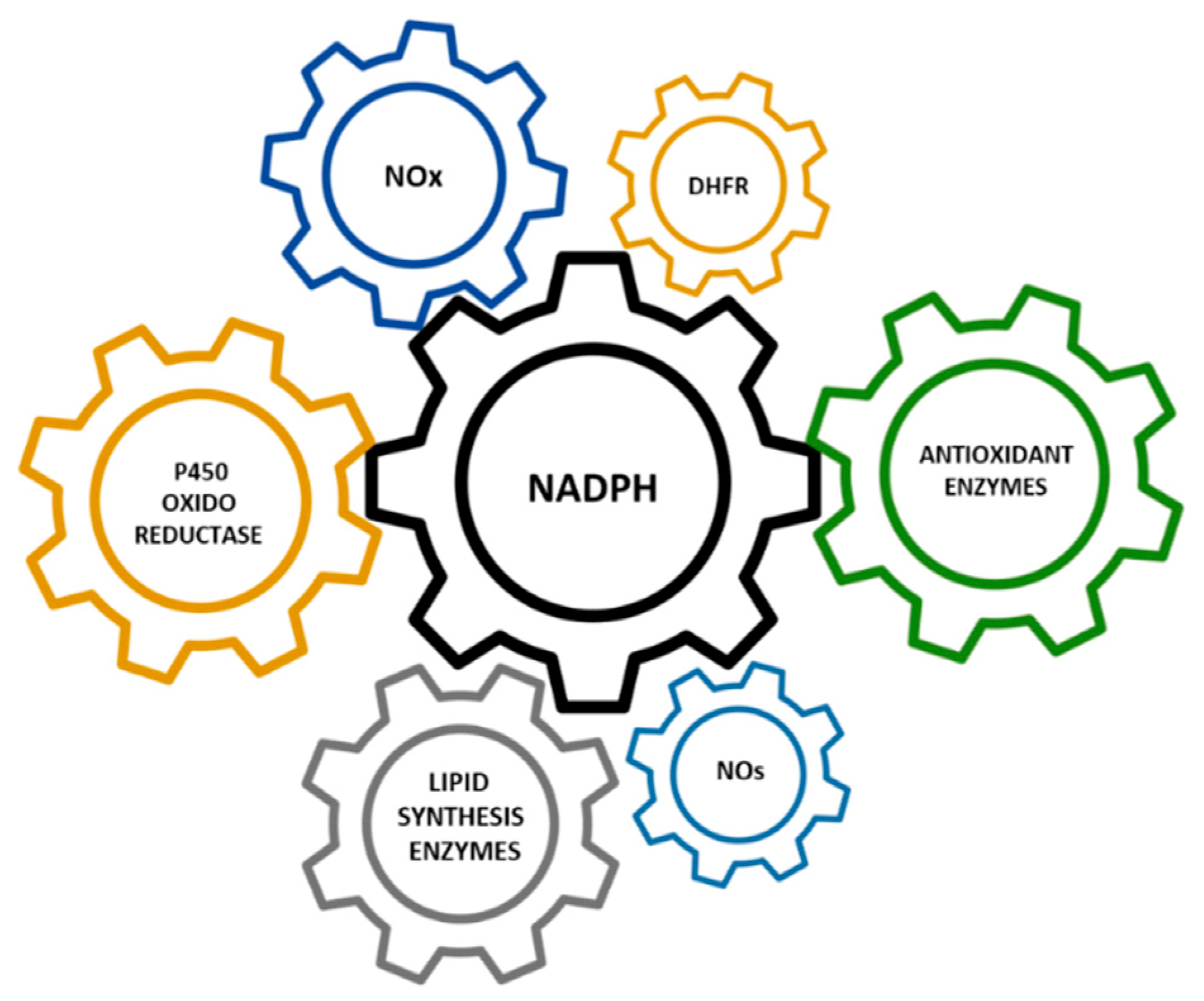
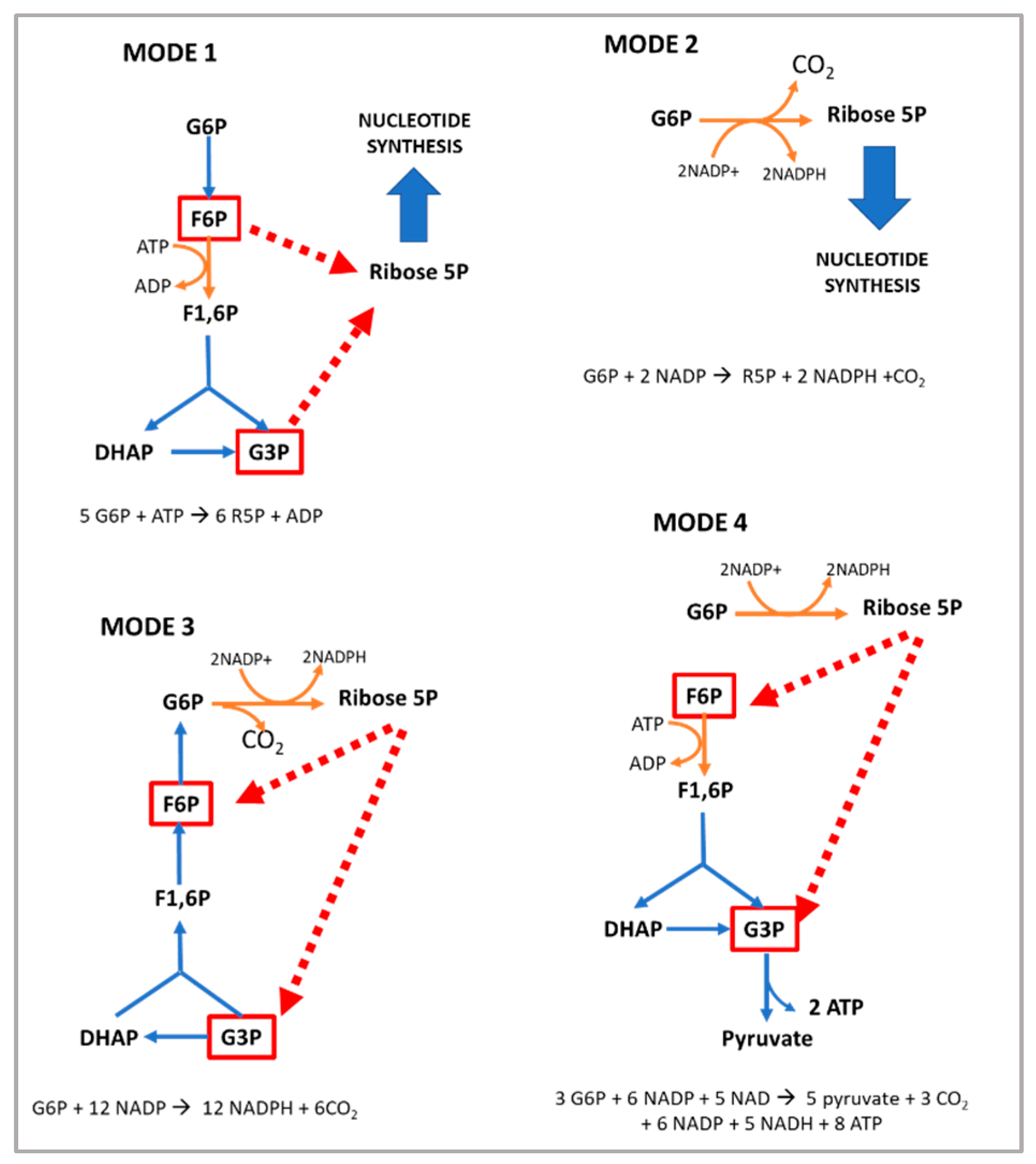
Hypomorphic Glucose 6-P dehydrogenase (G6PD) catalyzes the rate-limiting step in the pentose phosphate pathway (PPP), which provides the precursors of nucleotide synthesis for DNA replication as well as reduced nicotinamide adenine dinucleotide phosphate (NADPH). NADPH is involved in the detoxification of cellular reactive oxygen species (ROS) and de novo lipid synthesis. An association between increased PPP activity and the stimulation of cell growth has been reported in different tissues including the skeletal muscle, liver, and kidney. PPP activity is increased in skeletal muscle during embryogenesis, denervation, ischemia, mechanical overload, the injection of myonecrotic agents, and physical exercise. In fact, the highest relative increase in the activity of skeletal muscle enzymes after one bout of exhaustive exercise is that of G6PD, suggesting that the activation of the PPP occurs in skeletal muscle to provide substrates for muscle repair. The age-associated loss in muscle mass and strength leads to a decrease in G6PD activity and protein content in skeletal muscle. G6PD overexpression in Drosophila Melanogaster and mice protects against metabolic stress, oxidative damage, and age-associated functional decline, and results in an extended median lifespan.
1. The Pentose Phosphate Pathway and the Regulation of Glucose 6-P Dehydrogenase
2. Loss of Function Models for G6PD
3. G6PD and Cell Growth
The modulation of cell survival and cell growth relies on intracellular redox regulation [28]. As mentioned in the previous sections of this manuscript, NADPH—the principal intracellular reductant—is a critical modulator of redox potential. In 1999, Dr. Stanton and coworkers found that G6PD plays an important role in cell death by regulating intracellular redox levels [29]. The inhibition of G6PD by both dehydroepiandrosterone (DHEA) and 6-aminonicotinamide (6-ANAD) augmented cell death triggered by serum deprivation and oxidative stress, while the overexpression of G6PD in a cell line conferred resistance to H2O2-induced cell death. Previously, in G6PD-deficient cell lines, it was reported that these cells had decreased cloning efficiencies and growth rates and were highly sensitive to ROS when compared to cells expressing endogenous levels of the enzyme [30]. Consistent with these results, an association between the stimulation of cell growth in different tissues and increased PPP activity has also been reported [31]. Kidney hypertrophy due to unilateral nephrectomy is associated with increased G6PD activity [32], while the growth of rat liver cells stimulated by growth hormone is also associated with an increase in G6PD activity [33].
4. G6PD in the Regeneration of Skeletal Muscle after Damage
5. Positive Regulators of G6PD Activity in Skeletal Muscle
As shown in Table 1, G6PD can be regulated by pharmacological, nutritional, and physiological interventions, such as physical exercise [31].
| Positive Regulators | Negative Regulators |
|---|---|
| Acetylation [46] | 5′ adenosine monophosphate-activated protein kinase (AMPK) [47] |
| G6PD activator AG1 [48] | Aldosterone [49] |
| AKT [50] | Angiotensin II [51] |
| ATM serine/threonine kinase (ATM) [52] | Arachidonic acid [53] |
| Benfotiamine (vitamin B1 analog) [54][55] | Cyclic adenosine monophosphate (cAMP) [56] |
| Proto-oncogene tyrosine-protein kinase Src (c-Src) [57] | cAMP-dependent protein kinase A [56] |
| cGMP-dependent protein kinase G [58] | cAMP response element modulator (CREM) [49] |
| Cyclin D3-CDK6 [59] | Dehydroepiandrosterone (DHEA) [60] |
| Epidermal growth factor (EGF) [61] | miR-122 and miR-1 [62] |
| Estrogens [54] | p38 mitogen-activated protein kinase [53] |
| Exercise [23] | p53 [63] |
| Glycosylation [64] | Phosphatase and tensin homolog (PTEN) [65] |
| Growth hormone [54] | TP53 [63] |
| Hepatocyte growth factor (HGF) [66] | Tumor necrosis factor-α (TNFα) [31] |
| Heat shock protein 27 (Hsp27) [67] | |
| Hypoxia inducible factor (HIF) [68] | |
| Inhibitor of DNA binding 1 (ID1) [69] | |
| Insulin [70] | |
| Mammalian target of rapamycin (mTOR) [71] | |
| Nuclear-factor-E2-related factor (Nrf2) [72] | |
| Ribosomal protein S6 kinase beta-1 (p70S6K) [54] | |
| Serine/threonine-protein kinase PAK 4 (PAK4) [73] | |
| Protein disulfide isomerase family A, member 3 pseudogene (PDIA3P) [74] | |
| Phosphatidylinositol-3-kinase (PI-3K) [50] | |
| Phospholipase C [54] | |
| Phospholipase C-γ [75] | |
| Platelet-derived growth factor (PDGF) [75] | |
| Polo-like kinase 1 (PLK-1) [76] | |
| Ras-GTPase [31] | |
| S6 kinase [77] | |
| Snail [78] | |
| Sterol-responsive element bindingprotein (SREBP) 1 [31] | |
| Stobadine [79] | |
| TAp73 [80] | |
| Testosterone [54] | |
| Transforming growth factor beta 1 (TGF-β1) [81] | |
| TP53-induced glycolysis and apoptosis regulator (TIGAR) [82] | |
| Vascular endothelial cell growth factor (VEGF) [57] | |
| Vitamin D [83] | |
| Vitamin E [79] |
References
- Stincone, A.; Prigione, A.; Cramer, T.; Wamelink, M.M.; Campbell, K.; Cheung, E.; Olin-Sandoval, V.; Grüning, N.M.; Krüger, A.; Tauqeer Alam, M.; et al. The return of metabolism: Biochemistry and physiology of the pentose phosphate pathway. Biol. Rev. Camb. Philos. Soc. 2015, 90, 927–963.
- Baquer, N.Z.; Hothersall, J.S.; McLean, P. Function and regulation of the pentose phosphate pathway in brain. Curr. Top. Cell. Regul. 1988, 29, 265–289.
- Barcia-Vieitez, R.; Ramos-Martínez, J.I. The regulation of the oxidative phase of the pentose phosphate pathway: New answers to old problems. IUBMB Life 2014, 66, 775–779.
- Ghergurovich, J.M.; García-Cañaveras, J.C.; Wang, J.; Schmidt, E.; Zhang, Z.; TeSlaa, T.; Patel, H.; Chen, L.; Britt, E.C.; Piqueras-Nebot, M.; et al. A small molecule G6PD inhibitor reveals immune dependence on pentose phosphate pathway. Nat. Chem. Biol. 2020, 16, 731–739.
- Nóbrega-Pereira, S.; Fernandez-Marcos, P.J.; Brioche, T.; Gomez-Cabrera, M.C.; Salvador-Pascual, A.; Flores, J.M.; Viña, J.; Serrano, M. G6PD protects from oxidative damage and improves healthspan in mice. Nat. Commun. 2016, 7, 10894.
- Fernandez-Marcos, P.J.; Nobrega-Pereira, S. NADPH: New oxygen for the ROS theory of aging. Oncotarget 2016, 7, 50814–50815.
- Panday, A.; Sahoo, M.K.; Osorio, D.; Batra, S. NADPH oxidases: An overview from structure to innate immunity-associated pathologies. Cell. Mol. Immunol. 2015, 12, 5–23.
- Brown, D.I.; Griendling, K.K. Nox proteins in signal transduction. Free Radic. Biol. Med. 2009, 47, 1239–1253.
- Ferreira, L.F.; Laitano, O. Regulation of NADPH oxidases in skeletal muscle. Free Radic. Biol. Med. 2016, 98, 18–28.
- Berg, J.M.; Tymoczko, J.L.; Gatto, G.J., Jr.; Stryer, L.; Held, A.T.; Maxam, G.T.; Seidler, L.T.; Hacker, B.R.T.; Jarosch, B.T. Biochemistry, 8th ed.; Springer: Berlin/Heidelberg, Germany, 2017.
- Cho, E.S.; Cha, Y.H.; Kim, H.S.; Kim, N.H.; Yook, J.I. The Pentose Phosphate Pathway as a Potential Target for Cancer Therapy. Biomol. Ther. 2018, 26, 29–38.
- Eggleston, L.V.; Krebs, H.A. Regulation of the pentose phosphate cycle. Biochem. J. 1974, 138, 425–435.
- Salati, L.M.; Amir-Ahmady, B. Dietary regulation of expression of glucose-6-phosphate dehydrogenase. Annu. Rev. Nutr. 2001, 21, 121–140.
- Jiang, A.; Guo, H.; Jiang, X.; Tao, J.; Wu, W.; Liu, H. G6PD Deficiency is Crucial for Insulin Signaling Activation in Skeletal Muscle. Int. J. Mol. Sci. 2022, 23, 7425.
- Horton, J.D. Sterol regulatory element-binding proteins: Transcriptional activators of lipid synthesis. Biochem. Soc. Trans. 2002, 30, 1091–1095.
- Salati, L.M.; Szeszel-Fedorowicz, W.; Tao, H.; Gibson, M.A.; Amir-Ahmady, B.; Stabile, L.P.; Hodge, D.L. Nutritional regulation of mRNA processing. J. Nutr. 2004, 134, 2437S–2443S.
- Zhang, H.S.; Wang, S.Q. Nrf2 is involved in the effect of tanshinone IIA on intracellular redox status in human aortic smooth muscle cells. Biochem. Pharmacol. 2007, 73, 1358–1366.
- Marks, P.A.; Gross, R.T. Erythrocyte glucose-6-phosphate dehydrogenase deficiency: Evidence of differences between Negroes and Caucasians with respect to this genetically determined trait. J. Clin. Investig. 1959, 38, 2253–2262.
- Frank, J.E. Diagnosis and management of G6PD deficiency. Am. Fam. Physician 2005, 72, 1277–1282.
- Cappellini, M.D.; Fiorelli, G. Glucose-6-phosphate dehydrogenase deficiency. Lancet 2008, 371, 64–74.
- Ruwende, C.; Khoo, S.C.; Snow, R.W.; Yates, S.N.; Kwiatkowski, D.; Gupta, S.; Warn, P.; Allsopp, C.E.; Gilbert, S.C.; Peschu, N. Natural selection of hemi- and heterozygotes for G6PD deficiency in Africa by resistance to severe malaria. Nature 1995, 376, 246–249.
- Kletzien, R.F.; Harris, P.K.; Foellmi, L.A. Glucose-6-phosphate dehydrogenase: A “housekeeping” enzyme subject to tissue-specific regulation by hormones, nutrients, and oxidant stress. FASEB J. 1994, 8, 174–181.
- Arc-Chagnaud, C.; Salvador-Pascual, A.; Garcia-Dominguez, E.; Olaso-Gonzalez, G.; Correas, A.G.; Serna, E.; Brioche, T.; Chopard, A.; Fernandez-Marcos, P.J.; Serrano, M.; et al. Glucose 6-P dehydrogenase delays the onset of frailty by protecting against muscle damage. J. Cachexia Sarcopenia Muscle 2021, 12, 1879–1896.
- Luzzatto, L.; Arese, P. Favism and Glucose-6-Phosphate Dehydrogenase Deficiency. N. Engl. J. Med. 2018, 378, 1068–1069.
- Hecker, P.A.; Lionetti, V.; Ribeiro, R.F.; Rastogi, S.; Brown, B.H.; O’Connell, K.A.; Cox, J.W.; Shekar, K.C.; Gamble, D.M.; Sabbah, H.N.; et al. Glucose 6-phosphate dehydrogenase deficiency increases redox stress and moderately accelerates the development of heart failure. Circ. Heart Fail. 2013, 6, 118–126.
- Jeng, W.; Loniewska, M.M.; Wells, P.G. Brain glucose-6-phosphate dehydrogenase protects against endogenous oxidative DNA damage and neurodegeneration in aged mice. ACS Chem. Neurosci. 2013, 4, 1123–1132.
- Loniewska, M.M.; Gupta, A.; Bhatia, S.; MacKay-Clackett, I.; Jia, Z.; Wells, P.G. DNA damage and synaptic and behavioural disorders in glucose-6-phosphate dehydrogenase-deficient mice. Redox Biol. 2020, 28, 101332.
- Tian, W.N.; Braunstein, L.D.; Pang, J.; Stuhlmeier, K.M.; Xi, Q.C.; Tian, X.; Stanton, R.C. Importance of glucose-6-phosphate dehydrogenase activity for cell growth. J. Biol. Chem. 1998, 273, 10609–10617.
- Tian, W.N.; Braunstein, L.D.; Apse, K.; Pang, J.; Rose, M.; Tian, X.; Stanton, R.C. Importance of glucose-6-phosphate dehydrogenase activity in cell death. Am. J. Physiol. 1999, 276, C1121–C1131.
- Pandolfi, P.P.; Sonati, F.; Rivi, R.; Mason, P.; Grosveld, F.; Luzzatto, L. Targeted disruption of the housekeeping gene encoding glucose 6-phosphate dehydrogenase (G6PD): G6PD is dispensable for pentose synthesis but essential for defense against oxidative stress. EMBO J. 1995, 14, 5209–5215.
- Stanton, R.C. Glucose-6-phosphate dehydrogenase, NADPH, and cell survival. IUBMB Life 2012, 64, 362–369.
- Farquhar, J.K.; Scott, W.N.; Coe, F.L. Hexose monophosphate shunt activity in compensatory renal hypertrophy. Proc. Soc. Exp. Biol. Med. 1968, 129, 809–812.
- Schaffer, W.T. Effects of growth hormone on lipogenic enzyme activities in cultured rat hepatocytes. Am. J. Physiol. 1985, 248, E719–E725.
- Beaconsfield, P. Local metabolic response to physio-pathological demands: The pentose phosphate pathway. Experientia 1963, 19, 437–438.
- Beatty, C.H.; Peterson, R.D.; Basinger, G.M.; Bocek, R.M. Major metabolic pathways for carbohydrate metabolism of voluntary skeletal muscle. Am. J. Physiol. 1966, 210, 404–410.
- Wagner, K.R.; Kauffman, F.C.; Max, S.R. The pentose phosphate pathway in regenerating skeletal muscle. Biochem. J. 1978, 170, 17–22.
- Rifenberick, D.H.; Koski, C.L.; Max, S.R. Metabolic studies of skeletal muscle regeneration. Exp. Neurol. 1974, 45, 527–540.
- Smith, B. Histochemical changes in muscle necrosis and regeneration. J. Pathol. Bacteriol. 1965, 89, 139–143.
- Snow, M.H. Metabolic activity during the degenerative and early regenerative stages of minced skeletal muscle. Anat. Rec. 1973, 176, 185–203.
- Beaconsfield, P.; Carpi, A. Localization of an infectious lesion and glucose metabolism via the pentose phosphate pathway. Nature 1964, 201, 825–827.
- Boveris, A.; Erecinska, M.; Wagner, M. Reduction kinetics of cytochromes b. Biochim. Biophys. Acta 1972, 256, 223–242.
- Beaconsfield, P.; Reading, H.W. Pathways of glucose metabolism and nucleic acid synthesis. Nature 1964, 202, 464–466.
- Susheela, A.K.; Hudgson, P.; Walton, J.N. Murine muscular dystrophy. Some histochemical and biochemical observations. J. Neurol. Sci. 1968, 7, 437–463.
- Carlson, B.M. Regeneration of the rat gastrocnemius muscle from sibling and non-sibling muscle fragments. Am. J. Anat. 1970, 128, 21–31.
- Carlson, B.M. Relationship between the tissue and epimorphic regeneration of muscles. Am. Zool. 1970, 10, 175–186.
- Makarona, K.; Caputo, V.S.; Costa, J.R.; Liu, B.; O’Connor, D.; Iskander, D.; Roper, D.; Robertson, L.; Bhatnagar, N.; Terpos, E.; et al. Transcriptional and epigenetic basis for restoration of G6PD enzymatic activity in human G6PD-deficient cells. Blood 2014, 124, 134–141.
- Yang, L.; He, Z.; Yao, J.; Tan, R.; Zhu, Y.; Li, Z.; Guo, Q.; Wei, L. Regulation of AMPK-related glycolipid metabolism imbalances redox homeostasis and inhibits anchorage independent growth in human breast cancer cells. Redox Biol. 2018, 17, 180–191.
- Hwang, S.; Mruk, K.; Rahighi, S.; Raub, A.G.; Chen, C.H.; Dorn, L.E.; Horikoshi, N.; Wakatsuki, S.; Chen, J.K.; Mochly-Rosen, D. Correcting glucose-6-phosphate dehydrogenase deficiency with a small-molecule activator. Nat. Commun. 2018, 9, 4045.
- Leopold, J.A.; Dam, A.; Maron, B.A.; Scribner, A.W.; Liao, R.; Handy, D.E.; Stanton, R.C.; Pitt, B.; Loscalzo, J. Aldosterone impairs vascular reactivity by decreasing glucose-6-phosphate dehydrogenase activity. Nat. Med. 2007, 13, 189–197.
- Wagle, A.; Jivraj, S.; Garlock, G.L.; Stapleton, S.R. Insulin regulation of glucose-6-phosphate dehydrogenase gene expression is rapamycin-sensitive and requires phosphatidylinositol 3-kinase. J. Biol. Chem. 1998, 273, 14968–14974.
- Brioche, T.; Pagano, A.F.; Py, G.; Chopard, A. Muscle wasting and aging: Experimental models, fatty infiltrations, and prevention. Mol. Aspects Med. 2016, 50, 56–87.
- Cosentino, C.; Grieco, D.; Costanzo, V. ATM activates the pentose phosphate pathway promoting anti-oxidant defence and DNA repair. EMBO J. 2011, 30, 546–555.
- Talukdar, I.; Szeszel-Fedorowicz, W.; Salati, L.M. Arachidonic acid inhibits the insulin induction of glucose-6-phosphate dehydrogenase via p38 MAP kinase. J. Biol. Chem. 2005, 280, 40660–40667.
- Brioche, T.; Kireev, R.A.; Cuesta, S.; Gratas-Delamarche, A.; Tresguerres, J.A.; Gomez-Cabrera, M.C.; Viña, J. Growth hormone replacement therapy prevents sarcopenia by a dual mechanism: Improvement of protein balance and of antioxidant defenses. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2014, 69, 1186–1198.
- Katare, R.; Caporali, A.; Emanueli, C.; Madeddu, P. Benfotiamine improves functional recovery of the infarcted heart via activation of pro-survival G6PD/Akt signaling pathway and modulation of neurohormonal response. J. Mol. Cell. Cardiol. 2010, 49, 625–638.
- Zhang, Z.; Apse, K.; Pang, J.; Stanton, R.C. High glucose inhibits glucose-6-phosphate dehydrogenase via cAMP in aortic endothelial cells. J. Biol. Chem. 2000, 275, 40042–40047.
- Pan, S.; World, C.J.; Kovacs, C.J.; Berk, B.C. Glucose 6-phosphate dehydrogenase is regulated through c-Src-mediated tyrosine phosphorylation in endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 895–901.
- Patel, D.; Kandhi, S.; Kelly, M.; Neo, B.H.; Wolin, M.S. Dehydroepiandrosterone promotes pulmonary artery relaxation by NADPH oxidation-elicited subunit dimerization of protein kinase G 1α. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014, 306, L383–L391.
- Wang, H.; Nicolay, B.N.; Chick, J.M.; Gao, X.; Geng, Y.; Ren, H.; Gao, H.; Yang, G.; Williams, J.A.; Suski, J.M.; et al. The metabolic function of cyclin D3-CDK6 kinase in cancer cell survival. Nature 2017, 546, 426–430.
- Schwartz, A.G.; Pashko, L.L. Dehydroepiandrosterone, glucose-6-phosphate dehydrogenase, and longevity. Ageing Res. Rev. 2004, 3, 171–187.
- Tsao, M.S.; Earp, H.S.; Grisham, J.W. The effects of epidermal growth factor and the state of confluence on enzymatic activities of cultured rat liver epithelial cells. J. Cell. Physiol. 1986, 126, 167–173.
- Köberle, V.; Kronenberger, B.; Pleli, T.; Trojan, J.; Imelmann, E.; Peveling-Oberhag, J.; Welker, M.W.; Elhendawy, M.; Zeuzem, S.; Piiper, A.; et al. Serum microRNA-1 and microRNA-122 are prognostic markers in patients with hepatocellular carcinoma. Eur. J. Cancer 2013, 49, 3442–3449.
- Jiang, P.; Du, W.; Wang, X.; Mancuso, A.; Gao, X.; Wu, M.; Yang, X. p53 regulates biosynthesis through direct inactivation of glucose-6-phosphate dehydrogenase. Nat. Cell Biol. 2011, 13, 310–316.
- Rao, X.; Duan, X.; Mao, W.; Li, X.; Li, Z.; Li, Q.; Zheng, Z.; Xu, H.; Chen, M.; Wang, P.G.; et al. O-GlcNAcylation of G6PD promotes the pentose phosphate pathway and tumor growth. Nat. Commun. 2015, 6, 8468.
- Hong, X.; Song, R.; Song, H.; Zheng, T.; Wang, J.; Liang, Y.; Qi, S.; Lu, Z.; Song, X.; Jiang, H.; et al. PTEN antagonises Tcl1/hnRNPK-mediated G6PD pre-mRNA splicing which contributes to hepatocarcinogenesis. Gut 2014, 63, 1635–1647.
- Aird, K.M.; Worth, A.J.; Snyder, N.W.; Lee, J.V.; Sivanand, S.; Liu, Q.; Blair, I.A.; Wellen, K.E.; Zhang, R. ATM couples replication stress and metabolic reprogramming during cellular senescence. Cell Rep. 2015, 11, 893–901.
- Préville, X.; Salvemini, F.; Giraud, S.; Chaufour, S.; Paul, C.; Stepien, G.; Ursini, M.V.; Arrigo, A.P. Mammalian small stress proteins protect against oxidative stress through their ability to increase glucose-6-phosphate dehydrogenase activity and by maintaining optimal cellular detoxifying machinery. Exp. Cell Res. 1999, 247, 61–78.
- Yi, W.; Clark, P.M.; Mason, D.E.; Keenan, M.C.; Hill, C.; Goddard, W.A.; Peters, E.C.; Driggers, E.M.; Hsieh-Wilson, L.C. Phosphofructokinase 1 glycosylation regulates cell growth and metabolism. Science 2012, 337, 975–980.
- Yin, X.; Tang, B.; Li, J.H.; Wang, Y.; Zhang, L.; Xie, X.Y.; Zhang, B.H.; Qiu, S.J.; Wu, W.Z.; Ren, Z.G. ID1 promotes hepatocellular carcinoma proliferation and confers chemoresistance to oxaliplatin by activating pentose phosphate pathway. J. Exp. Clin. Cancer Res. 2017, 36, 166.
- Nakamura, T.; Yoshimoto, K.; Aoyama, K.; Ichihara, A. Hormonal regulations of glucose-6-phosphate dehydrogenase and lipogenesis in primary cultures of rat hepatocytes. J. Biochem. 1982, 91, 681–693.
- Tsouko, E.; Khan, A.S.; White, M.A.; Han, J.J.; Shi, Y.; Merchant, F.A.; Sharpe, M.A.; Xin, L.; Frigo, D.E. Regulation of the pentose phosphate pathway by an androgen receptor-mTOR-mediated mechanism and its role in prostate cancer cell growth. Oncogenesis 2014, 3, e103.
- Zimta, A.A.; Cenariu, D.; Irimie, A.; Magdo, L.; Nabavi, S.M.; Atanasov, A.G.; Berindan-Neagoe, I. The Role of Nrf2 Activity in Cancer Development and Progression. Cancers 2019, 11, 1755.
- Zhang, X.; Li, Y.; Shao, Y.; Xiao, J.; Zhu, G.; Li, F. PAK4 regulates G6PD activity by p53 degradation involving colon cancer cell growth. Cell Death Dis. 2017, 8, e2820.
- Yang, X.; Ye, H.; He, M.; Zhou, X.; Sun, N.; Guo, W.; Lin, X.; Huang, H.; Lin, Y.; Yao, R.; et al. LncRNA PDIA3P interacts with c-Myc to regulate cell proliferation via induction of pentose phosphate pathway in multiple myeloma. Biochem. Biophys. Res. Commun. 2018, 498, 207–213.
- Tian, W.N.; Pignatare, J.N.; Stanton, R.C. Signal transduction proteins that associate with the platelet-derived growth factor (PDGF) receptor mediate the PDGF-induced release of glucose-6-phosphate dehydrogenase from permeabilized cells. J. Biol. Chem. 1994, 269, 14798–14805.
- Ma, X.; Wang, L.; Huang, D.; Li, Y.; Yang, D.; Li, T.; Li, F.; Sun, L.; Wei, H.; He, K.; et al. Polo-like kinase 1 coordinates biosynthesis during cell cycle progression by directly activating pentose phosphate pathway. Nat. Commun. 2017, 8, 1506.
- Thakur, A.; Rahman, K.W.; Wu, J.; Bollig, A.; Biliran, H.; Lin, X.; Nassar, H.; Grignon, D.J.; Sarkar, F.H.; Liao, J.D. Aberrant expression of X-linked genes RbAp46, Rsk4, and Cldn2 in breast cancer. Mol. Cancer Res. 2007, 5, 171–181.
- Kim, N.H.; Cha, Y.H.; Lee, J.; Lee, S.H.; Yang, J.H.; Yun, J.S.; Cho, E.S.; Zhang, X.; Nam, M.; Kim, N.; et al. Snail reprograms glucose metabolism by repressing phosphofructokinase PFKP allowing cancer cell survival under metabolic stress. Nat. Commun. 2017, 8, 14374.
- Ulusu, N.N.; Sahilli, M.; Avci, A.; Canbolat, O.; Ozansoy, G.; Ari, N.; Bali, M.; Stefek, M.; Stolc, S.; Gajdosik, A.; et al. Pentose phosphate pathway, glutathione-dependent enzymes and antioxidant defense during oxidative stress in diabetic rodent brain and peripheral organs: Effects of stobadine and vitamin E. Neurochem. Res. 2003, 28, 815–823.
- Du, W.; Jiang, P.; Mancuso, A.; Stonestrom, A.; Brewer, M.D.; Minn, A.J.; Mak, T.W.; Wu, M.; Yang, X. TAp73 enhances the pentose phosphate pathway and supports cell proliferation. Nat. Cell Biol. 2013, 15, 991–1000.
- Zhang, R.; Tao, F.; Ruan, S.; Hu, M.; Hu, Y.; Fang, Z.; Mei, L.; Gong, C. The TGFβ1-FOXM1-HMGA1-TGFβ1 positive feedback loop increases the cisplatin resistance of non-small cell lung cancer by inducing G6PD expression. Am. J. Transl. Res. 2019, 11, 6860–6876.
- Wang, J.; Duan, Z.; Nugent, Z.; Zou, J.X.; Borowsky, A.D.; Zhang, Y.; Tepper, C.G.; Li, J.J.; Fiehn, O.; Xu, J.; et al. Reprogramming metabolism by histone methyltransferase NSD2 drives endocrine resistance via coordinated activation of pentose phosphate pathway enzymes. Cancer Lett. 2016, 378, 69–79.
- Sardar, S.; Chakraborty, A.; Chatterjee, M. Comparative effectiveness of vitamin D3 and dietary vitamin E on peroxidation of lipids and enzymes of the hepatic antioxidant system in Sprague—Dawley rats. Int. J. Vitam. Nutr. Res. 1996, 66, 39–45.
- Schulpis, K.H.; Reclos, G.J.; Parthimos, T.; Parthimos, N.; Gavriilidis, A.; Tsakiris, S. L-cysteine supplementation protects the erythrocyte glucose-6-phosphate dehydrogenase activity from reduction induced by forced training. Clin. Biochem. 2006, 39, 1002–1006.
- Tsakiris, S.; Reclos, G.J.; Parthimos, T.; Tsakiris, T.; Parthimos, N.; Schulpis, K.H. α-Tocopherol supplementation restores the reduction of erythrocyte glucose-6-phosphate dehydrogenase activity induced by forced training. Pharmacol. Res. 2006, 54, 373–379.
- Schulpis, K.H.; Tsironi, M.; Skenderi, K.; Lazaropoulou, C.; Parthimos, N.; Reclos, G.; Goussetis, E.; Tsakiris, S.; Papassotiriou, I. Dramatic reduction of erythrocyte glucose-6-phosphate dehydrogenase activity in athletes participating in the ultradistance foot race “Spartathlon”. Scand. J. Clin. Lab. Investig. 2008, 68, 228–232.
- Tsakiris, S.; Parthimos, T.; Reclos, G.J.; Parthimos, N.; Tsakiris, T.; Schulpis, K.H. Significant reduction of erythrocyte glucose-6-phosphate dehydrogenase activity in soccer-players during play. Evidence for catecholamine mediated enzyme inhibition. Clin. Chem. Lab. Med. 2009, 47, 621–624.
- Boström, S.; Fahlén, M.; Hjalmarson, A.; Johansson, R. Activities of rat muscle enzymes after acute exercise. Acta Physiol. Scand. 1974, 90, 544–554.
- Schwane, J.A.; Armstrong, R.B. Effect of training on skeletal muscle injury from downhill running in rats. J. Appl. Physiol. Respir. Environ. Exerc. Physiol. 1983, 55, 969–975.
- Valentino, T.; Figueiredo, V.C.; Mobley, C.B.; McCarthy, J.J.; Vechetti, I.J. Evidence of myomiR regulation of the pentose phosphate pathway during mechanical load-induced hypertrophy. Physiol. Rep. 2021, 9, e15137.
- Turner, L.V.; Manchester, K.L. Glucose and glycogen metabolism in hypertrophied denervated rat hemidiaphragm. Biochem. J. 1970, 117, 33P.
- Weyrauch, L.A.; McMillin, S.L.; Witczak, C.A. Insulin Resistance Does Not Impair Mechanical Overload-Stimulated Glucose Uptake, but Does Alter the Metabolic Fate of Glucose in Mouse Muscle. Int. J. Mol. Sci. 2020, 21, 4715.
- Place, N.; Ivarsson, N.; Venckunas, T.; Neyroud, D.; Brazaitis, M.; Cheng, A.J.; Ochala, J.; Kamandulis, S.; Girard, S.; Volungevicius, G.; et al. Ryanodine receptor fragmentation and sarcoplasmic reticulum Ca2+ leak after one session of high-intensity interval exercise. Proc. Natl. Acad. Sci. USA 2015, 112, 15492–15497.
- Ristow, M.; Zarse, K.; Oberbach, A.; Kloting, N.; Birringer, M.; Kiehntopf, M.; Stumvoll, M.; Kahn, C.R.; Bluher, M. Antioxidants prevent health-promoting effects of physical exercise in humans. Proc. Natl. Acad. Sci. USA 2009, 106, 8665–8670.
- Paulsen, G.; Cumming, K.T.; Holden, G.; Hallen, J.; Ronnestad, B.R.; Sveen, O.; Skaug, A.; Paur, I.; Bastani, N.E.; Ostgaard, H.N.; et al. Vitamin C and E supplementation hampers cellular adaptation to endurance training in humans: A double-blind, randomised, controlled trial. J. Physiol. 2014, 592, 1887–1901.
- Gomez-Cabrera, M.C.; Domenech, E.; Romagnoli, M.; Arduini, A.; Borras, C.; Pallardo, F.V.; Sastre, J.; Vina, J. Oral administration of vitamin C decreases muscle mitochondrial biogenesis and hampers training-induced adaptations in endurance performance. Am. J. Clin. Nutr. 2008, 87, 142–149.
- Melikoglu, M.A.; Kaldirimci, M.; Katkat, D.; Sen, I.; Kaplan, I.; Senel, K. The effect of regular long term training on antioxidant enzymatic activities. J. Sports Med. Phys. Fit. 2008, 48, 388–390.
- Spodaryk, K.; Szyguła, Z.; Dabrowski, Z.; Miszta, H. The activity of erythrocyte enzymes in rats subjected to running exercises. Eur. J. Appl. Physiol. Occup. Physiol. 1985, 54, 533–537.
- Gomez-Cabrera, M.C.; Borrás, C.; Pallardó, F.V.; Sastre, J.; Ji, L.L.; Viña, J. Decreasing xanthine oxidase-mediated oxidative stress prevents useful cellular adaptations to exercise in rats. J. Physiol. 2005, 567, 113–120.
- Gomez-Cabrera, M.C.; Domenech, E.; Viña, J. Moderate exercise is an antioxidant: Upregulation of antioxidant genes by training. Free Radic. Biol. Med. 2008, 44, 126–131.
- Herscovich, S.; Gershon, D. Effects of aging and physical training on the neuromuscular junction of the mouse. Gerontology 1987, 33, 7–13.
- Griffiths, M.A.; Baker, D.H.; Novakofski, J.E.; Ji, L.L. Effects of exercise training on diet-induced lipogenic enzymes and body composition in rats. J. Am Coll. Nutr. 1993, 12, 155–161.
- Pereira, B.; Costa Rosa, L.F.; Safi, D.A.; Medeiros, M.H.; Curi, R.; Bechara, E.J. Superoxide dismutase, catalase, and glutathione peroxidase activities in muscle and lymphoid organs of sedentary and exercise-trained rats. Physiol. Behav. 1994, 56, 1095–1099.
- Borges-Silva, C.N.; Fonseca-Alaniz, M.H.; Alonso-Vale, M.I.; Takada, J.; Andreotti, S.; Peres, S.B.; Cipolla-Neto, J.; Pithon-Curi, T.C.; Lima, F.B. Reduced lipolysis and increased lipogenesis in adipose tissue from pinealectomized rats adapted to training. J. Pineal. Res. 2005, 39, 178–184.
- Ninfali, P.; Bresolin, N. Muscle glucose 6-phosphate dehydrogenase (G6PD) deficiency and oxidant stress during physical exercise. Cell Biochem. Funct. 1995, 13, 297–298.
- Jamurtas, A.Z.; Fatouros, I.G.; Deli, C.K.; Georgakouli, K.; Poulios, A.; Draganidis, D.; Papanikolaou, K.; Tsimeas, P.; Chatzinikolaou, A.; Avloniti, A.; et al. The Effects of Acute Low-Volume HIIT and Aerobic Exercise on Leukocyte Count and Redox Status. J. Sports Sci. Med. 2018, 17, 501–508.
- Georgakouli, K.; Fatouros, I.G.; Draganidis, D.; Papanikolaou, K.; Tsimeas, P.; Deli, C.K.; Jamurtas, A.Z. Exercise in Glucose-6-Phosphate Dehydrogenase Deficiency: Harmful or Harmless? A Narrative Review. Oxid. Med. Cell. Longev. 2019, 2019, 8060193.
- Jamurtas, A.Z.; Fatouros, I.G.; Koukosias, N.; Manthou, E.; Tofas, T.; Yfanti, C.; Nikolaidis, M.G.; Koutedakis, Y. Effect of exercise on oxidative stress in individuals with glucose-6-phosphate dehydrogenase deficiency. In Vivo 2006, 20, 875–880.
- Theodorou, A.A.; Nikolaidis, M.G.; Paschalis, V.; Sakellariou, G.K.; Fatouros, I.G.; Koutedakis, Y.; Jamurtas, A.Z. Comparison between glucose-6-phosphate dehydrogenase-deficient and normal individuals after eccentric exercise. Med. Sci. Sports Exerc. 2010, 42, 1113–1121.


