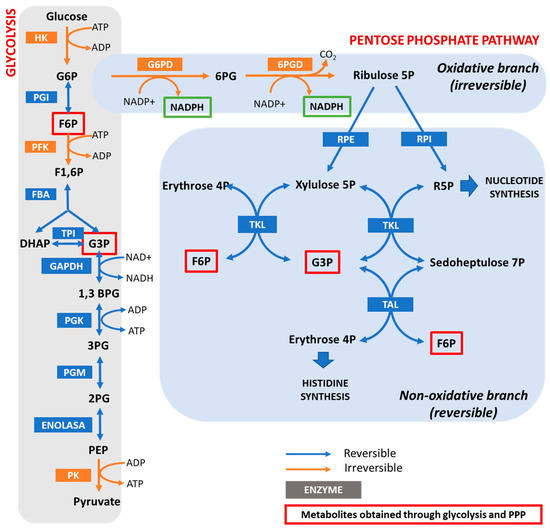
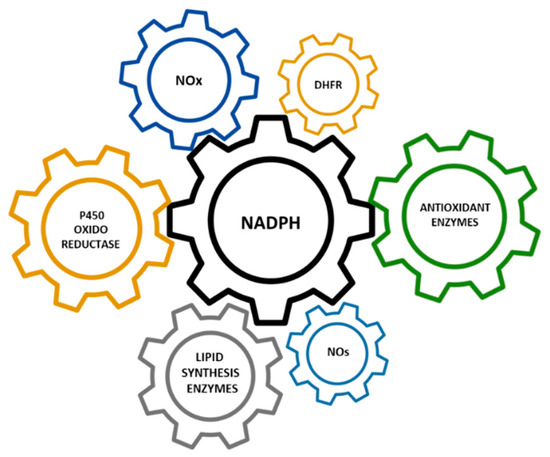
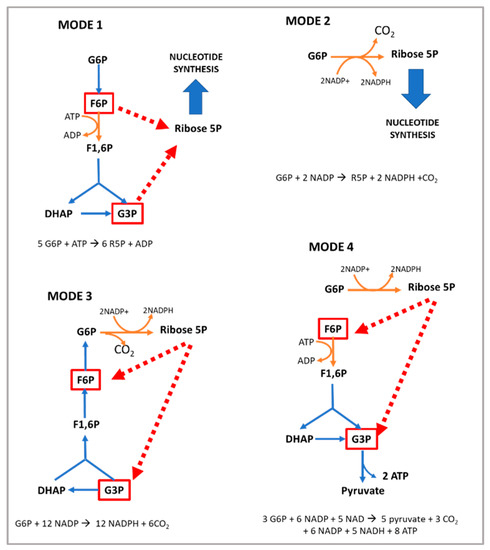
G6PD is the key enzyme in the regulation of the PPP. Therefore, any factor able to modify the level or activity of G6PD will determine the flow of the PPP. The “coarse control” of the PPP is carried out by modifications in the levels, location, and activity of G6PD [
16]. Factors such as diet composition can induce changes in the synthesis of G6PD [
17,
18]. An excess of carbohydrates in the diet leads to lipogenesis and the deposition of fat, which is associated with a 5–10-fold increase in G6PD activity in the liver [
16]. Accordingly, the expression of the G6PD gene is upregulated by the major transcription factor sterol-responsive element binding protein [
19]. High insulin and low glucagon levels, which are associated with this kind of diet, have also been described as regulators of G6PD through the control of mRNA synthesis [
20]. The transcription factor Nrf2, which regulates the antioxidant cellular response, also enhances G6PD gene expression [
21]. Interestingly, diets rich in polyunsaturated fatty acids (PUFAs) have the opposite effect on G6PD levels.
2. Loss of Function Models for G6PD
G6PD deficiency is the most common human enzymopathy. It is very heterogeneous and was first described in humans by Marks and Gross in 1959 [
26]. Approximately 400 million people worldwide carry a mutation in the G6PD gene, which causes an enzyme deficiency. Deficient alleles are prevalent in South and North America and in northern Europe [
27]. However, the highest prevalence of this enzymopathy is reported in Africa, the Middle East, the central and southern Pacific Islands, southern Europe, and southeast Asia. The global distribution of the G6PD deficiency is strikingly similar to that of malaria. In areas where G6PD deficiency is common,
Plasmodium Falciparum malaria is endemic, supporting the so-called malaria protection hypothesis [
28]. Epidemiological evidence for the association between G6PD deficiency and a reduction in the risk of severe malaria [
29] has been accompanied by the results of in vitro work showing that parasite growth is slowest in G6PD-deficient cells [
28].
The G6PD gene is located at the telomeric region in the X chromosome. Thus, its deficiency is an X-linked hereditary defect that causes variants with different clinical phenotypes (about 140 mutations have been described). The G6PD-encoding gene has been well preserved throughout evolution [
31]. As a monomer, the protein is inactive; however, as a dimer or tetramer, it is active. In its catalytic center, there is an amino acid sequence that binds to NADPH. The deficiency is caused by protein instability due to amino acid substitutions in different enzyme locations [
28]. The diagnosis of G6PD deficiency is based on the spectrophotometric quantification of the enzyme’s activity [
32].
The most frequent clinical manifestations of G6PD deficiency are acute and chronic hemolytic anemia and neonatal jaundice [
28]. The prevention of hemolysis by avoiding oxidative stress represents the most effective management of G6PD deficiency. Oxidative stress can be triggered by agents such as drugs (primaquine, sulfonamide, or acetanilide), infections (hepatitis viruses, cytomegalovirus, or pneumonia), or the ingestion of fava beans (favism). Favism is a hemolytic response to the consumption of fava beans that takes place in some individuals with G6PD deficiency [
33]. Isouramil, divicine, and convicine are thought to be the toxic constituents of fava beans that lead to the onset of the clinical manifestations of deficiency [
28]. The mechanism by which increased sensitivity to oxidative damage leads to hemolysis has not been fully elucidated [
34].
The risk of redox-mediated damage to brain cells in G6PD deficiency has also been studied [
63]. G6PD is an important enzyme in the protection against age-associated ROS neurodegenerative effects, and more specifically in the age-associated increase in oxidative DNA damage in the brain [
63]. Recently, brain damage associated with ROS production in G6PD-deficient animals was also found to have functional consequences. Old G6PD-deficient male mice exhibited synaptic dysfunction in their hippocampal slices while young and old G6PD-deficient females exhibited deficits in executive functions and social dominance [
64].
3. G6PD and Cell Growth
The modulation of cell survival and cell growth relies on intracellular redox regulation [65]. As mentioned in the previous sections of this manuscript, NADPH—the principal intracellular reductant—is a critical modulator of redox potential. In 1999, Dr. Stanton and coworkers found that G6PD plays an important role in cell death by regulating intracellular redox levels [66]. The inhibition of G6PD by both dehydroepiandrosterone (DHEA) and 6-aminonicotinamide (6-ANAD) augmented cell death triggered by serum deprivation and oxidative stress, while the overexpression of G6PD in a cell line conferred resistance to H2O2-induced cell death. Previously, in G6PD-deficient cell lines, it was reported that these cells had decreased cloning efficiencies and growth rates and were highly sensitive to ROS when compared to cells expressing endogenous levels of the enzyme [67]. Consistent with these results, an association between the stimulation of cell growth in different tissues and increased PPP activity has also been reported [68]. Kidney hypertrophy due to unilateral nephrectomy is associated with increased G6PD activity [69], while the growth of rat liver cells stimulated by growth hormone is also associated with an increase in G6PD activity [70].
4. G6PD in the Regeneration of Skeletal Muscle after Damage
The hexose monophosphate shunt is considered an almost negligible pathway in normal muscle. For this reason, the function of G6PD in skeletal muscle has been poorly investigated.
In vitro studies have shown that, under normal conditions, glucose breakdown takes place via both the Embden–Meyerhof pathway and the PPP in the liver, pancreas, arterial wall, kidney, spleen, and adrenals. However, in the central nervous system and cardiac and striated muscle, it is metabolized mainly via the glycolytic route [
72]. In addition, several conditions increase the activity of the PPP in skeletal muscle: (i) embryogenesis [
73]; (ii) denervation; (iii) ischemia; (iv) hypertrophy; (v) the injection of myonecrotic agents with local degeneration effects [
74,
75]; and (vi) physical exercise [
32].
The injection of myonecrotic agents (bupivacaine, Marcaine, or cardiotoxin) induces a rapid (8 h) and dramatic (6–9-fold) increase in the activities of G6PD and 6PGD during regeneration after muscle destruction. By using histological techniques [
76,
77], it has been shown that G6PD is localized within muscle cells in regenerating muscle; thus, the enhanced enzyme activity resides in the muscle fibers themselves for at least the first 6–8 h after Marcaine injection. After that time, phagocytic cells contribute to the increase in enzyme activity [
74]. The enhanced activities of G6PD and 6PGD likely reflect accelerated glucose utilization for the production of nucleic acids and lipids [
75,
78,
79,
80]. In this regard, increased quantities of RNA have been noted in a number of studies on muscle regeneration [
81,
82,
83]. The enhancement of the PPP is important for anabolic processes in the initial stages of skeletal muscle regeneration; however, the role of G6PD in skeletal muscle goes beyond biosynthetic processes.
5. Positive Regulators of G6PD Activity in Skeletal Muscle
As shown in Table 2, G6PD can be regulated by pharmacological, nutritional, and physiological interventions, such as physical exercise [68].
Table 2. Cellular signals regulating G6PD and the PPP.
| Positive Regulators |
Negative Regulators |
| Acetylation [130] |
5′ adenosine monophosphate-activated protein kinase (AMPK) [131] |
| G6PD activator AG1 [132] |
Aldosterone [133] |
| AKT [134] |
Angiotensin II [120] |
| ATM serine/threonine kinase (ATM) [135] |
Arachidonic acid [136] |
| Benfotiamine (vitamin B1 analog) [110,137] |
Cyclic adenosine monophosphate (cAMP) [138] |
| Proto-oncogene tyrosine-protein kinase Src (c-Src) [25] |
cAMP-dependent protein kinase A [138] |
| cGMP-dependent protein kinase G [139] |
cAMP response element modulator (CREM) [133] |
| Cyclin D3-CDK6 [140] |
Dehydroepiandrosterone (DHEA) [141] |
| Epidermal growth factor (EGF) [142] |
miR-122 and miR-1 [143] |
| Estrogens [110] |
p38 mitogen-activated protein kinase [136] |
| Exercise [32] |
p53 [144] |
| Glycosylation [145] |
Phosphatase and tensin homolog (PTEN) [146] |
| Growth hormone [110] |
TP53 [144] |
| Hepatocyte growth factor (HGF) [147] |
Tumor necrosis factor-α (TNFα) [68] |
| Heat shock protein 27 (Hsp27) [148] |
|
| Hypoxia inducible factor (HIF) [149] |
|
| Inhibitor of DNA binding 1 (ID1) [150] |
|
| Insulin [151] |
|
| Mammalian target of rapamycin (mTOR) [152] |
|
| Nuclear-factor-E2-related factor (Nrf2) [153] |
|
| Ribosomal protein S6 kinase beta-1 (p70S6K) [110] |
|
| Serine/threonine-protein kinase PAK 4 (PAK4) [154] |
|
| Protein disulfide isomerase family A, member 3 pseudogene (PDIA3P) [155] |
|
| Phosphatidylinositol-3-kinase (PI-3K) [134] |
|
| Phospholipase C [110] |
|
| Phospholipase C-γ [156] |
|
| Platelet-derived growth factor (PDGF) [156] |
|
| Polo-like kinase 1 (PLK-1) [157] |
|
| Ras-GTPase [68] |
|
| S6 kinase [158] |
|
| Snail [159] |
|
| Sterol-responsive element bindingprotein (SREBP) 1 [68] |
|
| Stobadine [160] |
|
| TAp73 [161] |
|
| Testosterone [110] |
|
| Transforming growth factor beta 1 (TGF-β1) [162] |
|
| TP53-induced glycolysis and apoptosis regulator (TIGAR) [163] |
|
| Vascular endothelial cell growth factor (VEGF) [25] |
|
| Vitamin D [164] |
|
| Vitamin E [160] |
|
G6PD activity has been studied in both skeletal muscle and erythrocytes after one bout of exhaustive exercise. Surprisingly, contradictory results were found in the literature.
G6PD activity in erythrocytes is reduced in humans after one bout of high intensity exercise (~40%), likely due to ROS generation [
89]. Accordingly, supplementation with L-cysteine for a week (0.5 g/24 h) [
89] or with α-Tocopherol for a month (200 mg/24 h) [
90] leads to the maintenance of the enzyme activity. These results have also been verified in long distance runners [
91] and soccer players [
91,
92]. To the contrary, the highest relative increase in enzyme activities, both for mitochondrial and extramitochondrial enzymes, after exhaustive swimming in rat skeletal muscle was shown for G6PD and 6PGD, which increased by 115% and 40%, respectively, 1 and 3 days after an acute bout of exercise [
93]. Similarly, an increase in muscle G6PD activity of ~100–350% was observed after a downhill running protocol in rats [
94], suggesting that the activation of the PPP occurs in skeletal muscle to provide substrates for muscle repair.
The PPP has been proven to be a fundamental metabolic pathway that allows for rapid and robust hypertrophic growth in muscle cells in response to mechanical overload [
99]. For example, the denervation of one half of the diaphragm was shown to induce transient hypertrophy in the muscle on the other side [
100]. In this model, the activities of G6PD and 6PDG increased immediately after denervation, reaching a maximum after 3 days [
100]. More recently, the importance of G6PD in the regulation of skeletal muscle metabolism during hypertrophy was highlighted in a study analyzing gene expression from a transcriptomic microarray of specific metabolic pathways in mechanically overloaded plantaris muscle-induced hypertrophy [
99]. A robust increase in G6PD mRNA expression was found in the overloaded muscle throughout the whole analyzed time course (1, 3, 5, and 7 days), consistent with an increase in NADPH levels to support nucleotide biosynthesis and to boost the muscle antioxidant defense [
99]. It was also shown that the abundance of the G6PD protein significantly increased (~140%) in response to 5 days of mechanical overload in muscle [
101].
Physical exercise acutely increases ROS generation; however, if practiced regularly, it induces positive adaptations in mitochondrial density [
112,
113,
114,
115] and antioxidant defenses, including increased G6PD enzymatic activity [
115,
116,
117]. Growing evidence suggests that physical training upregulates the level of antioxidant enzymes in the tissues actively involved in exercise [
118,
119].
The age-associated loss in muscle mass and strength (i.e., sarcopenia) leads to a decrease in G6PD activity and protein content in skeletal muscle [
120]. Whether the well-known positive effects of exercise training in old individuals are mediated through an increase in G6PD activity should be further studied in depth. The results published to date are contradictory and do not allow definitive conclusions to be drawn [
121,
122,
123,
124].
Finally, the question of whether exercise training is a safe and useful intervention in G6PD-deficient patients is something that has been an object of debate. G6PD-deficient individuals, as previously mentioned, are less protected against oxidative stress and could be predisposed to oxidative damage when they perform high-intensity physical training [
125]. However, several studies have shown that exercise intensity does not cause oxidative stress or hemolysis above those levels expected in people without G6PD deficiency [
126,
127,
128,
129]. Therefore, despite the limited published studies, it seems that G6PD-deficient patients can safely participate in physical exercise programs with different intensities and durations.