 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Clint Gray | -- | 3618 | 2022-11-01 04:14:44 | | | |
| 2 | Jessie Wu | Meta information modification | 3618 | 2022-11-01 06:38:02 | | | | |
| 3 | Jessie Wu | -6 word(s) | 3612 | 2022-11-01 06:40:31 | | |
Video Upload Options
Fructose is a 6-carbon polyhydroxy ketone monosaccharide that shares the same chemical formula and is an isomer of glucose. Fructose consumption is now recognised as a major risk factor in the development of metabolic diseases, such as hyperlipidaemia, diabetes, non-alcoholic fatty liver disease and obesity. In addition to environmental, social, and genetic factors, an unfavourable intrauterine environment is now also recognised as an important factor in the progression of, or susceptibility to, metabolic disease during adulthood. Developmental trajectory in the short term, in response to nutrient restriction or excessive nutrient availability, may promote adaptation that serves to maintain organ functionality necessary for immediate survival and foetal development. Consequently, this may lead to decreased function of organ systems when presented with an unfavourable neonatal, adolescent and/or adult nutritional environment. These early events may exacerbate susceptibility to later-life disease since sub-optimal maternal nutrition increases the risk of non-communicable diseases (NCDs) in future generations. Earlier dietary interventions, implemented in pregnant mothers or those considering pregnancy, may have added benefit.
1. Fructose Metabolism
2. Hepatocyte Metabolism of Fructose
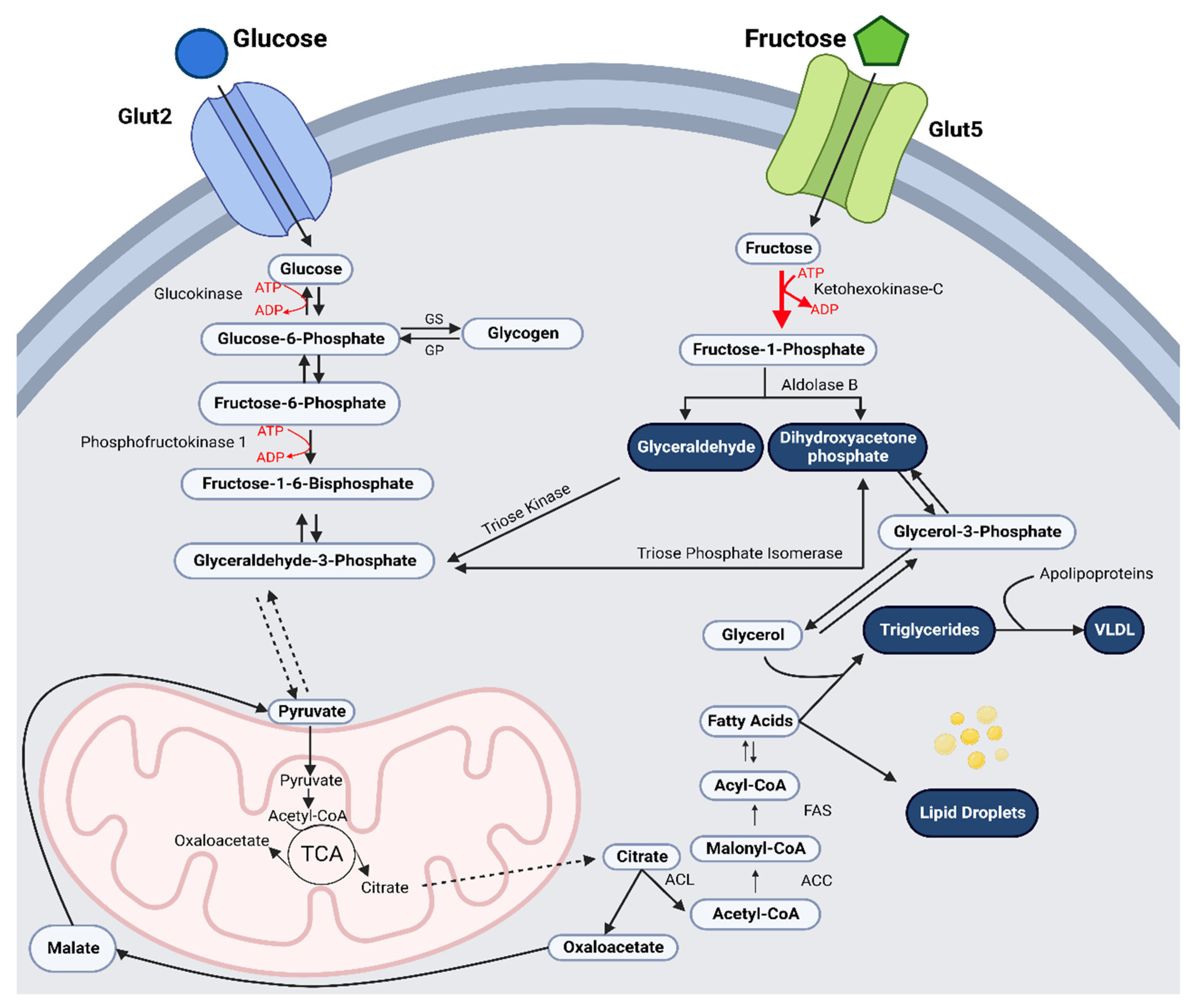
3. Excess Fructose and Hepatic De Novo Lipogenesis and Triglyceride Synthesis
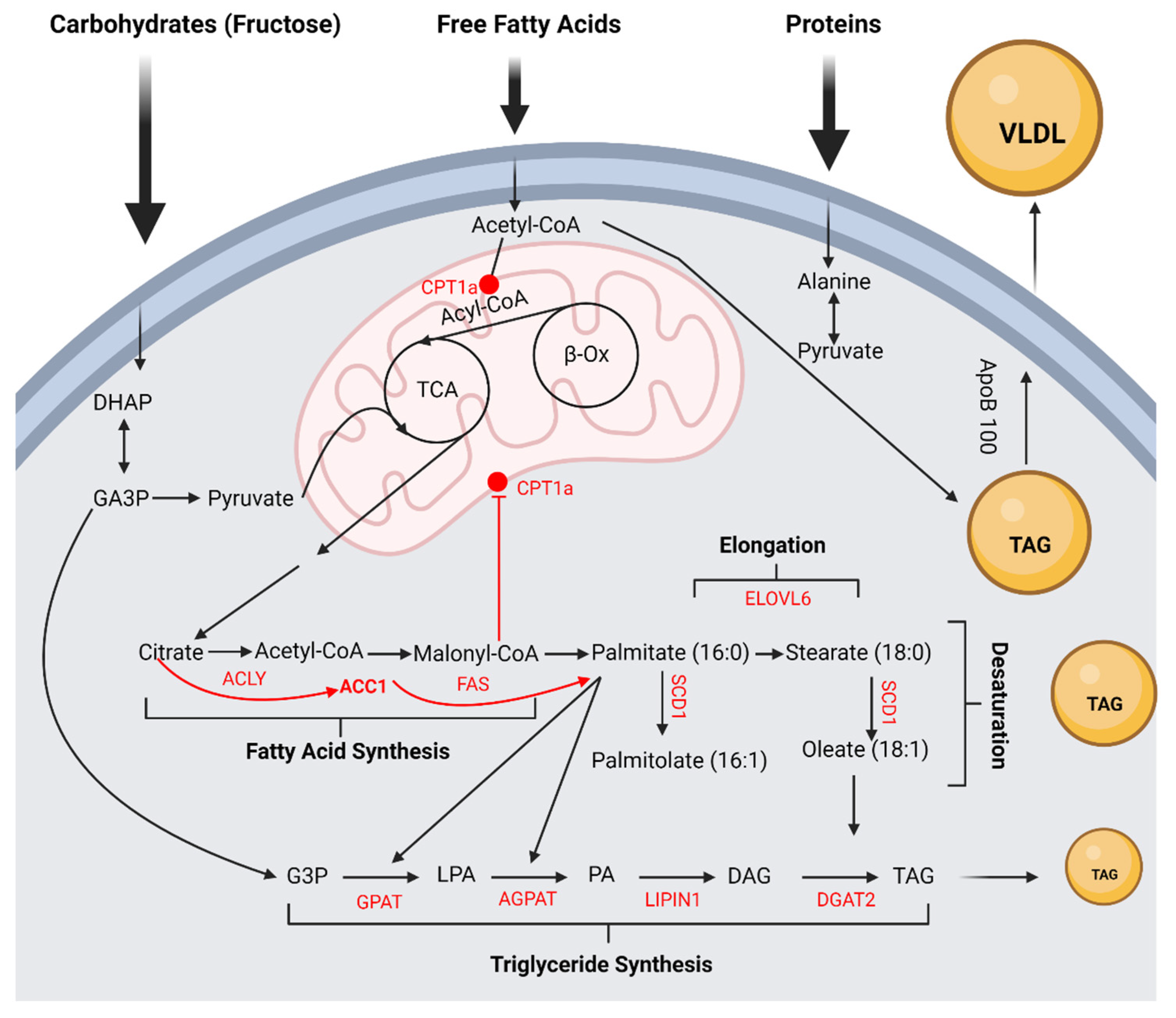
4. Adverse Effects of Excessive Fructose Intake
5. Excess Maternal Fructose Intake and Offspring Predisposition to Metabolic Dysfunction
References
- Biro, G. Human biological characteristics of fructose. J. Food Investig. 2018, 64, 1908–1917.
- Zhang, D.-M.; Jiao, R.-Q.; Kong, L.-D. High Dietary Fructose: Direct or Indirect Dangerous Factors Disturbing Tissue and Organ Functions. Nutrients 2017, 9, 335.
- Sun, S.Z.; Empie, M.W. Fructose metabolism in humans—What isotopic tracer studies tell us. Nutr. Metab. 2012, 9, 89.
- Douard, V.; Ferraris, R.P. Regulation of the fructose transporter GLUT5 in health and disease. Am. J. Physiol. Endocrinol. Metab. 2008, 295, E227–E237.
- Ferraris, R.P.; Choe, J.Y.; Patel, C.R. Intestinal absorption of fructose. Annu. Rev. Nutr. 2018, 38, 41–67.
- Ter Horst, K.W.; Serlie, M.J. Fructose Consumption, Lipogenesis, and Non-Alcoholic Fatty Liver Disease. Nutrients 2017, 9, 981.
- Ebert, K.; Ludwig, M.; Geillinger, K.E.; Schoberth, G.C.; Essenwanger, J.; Stolz, J.; Daniel, H.; Witt, H. Reassessment of GLUT7 and GLUT9 as Putative Fructose and Glucose Transporters. J. Membr. Biol. 2017, 250, 171–182.
- Dekker, M.J.; Qiaozhu, S.; Baker, C.; Rutledge, A.C.; Adeli, K. Fructose: A highly lipogenic nutrient implicated in insulin resistance, hepatic steatosis, and the metabolic syndrome Mechanism of liponecrosis, a distinct mode of programmed cell death View project. Artic. AJP Endocrinol. Metab. 2010, 299, E685–E694.
- Baynes, J.W.; Dominiczak, M.H. Medical Biochemistry, 2nd ed.; Elsevier Mosby: Orlando, FL, USA, 2005; p. 693.
- Regnault, T.R.; Gentili, S.; Sarr, O.; Toop, C.R.; Sloboda, D.M. Fructose, pregnancy and later life impacts. Clin. Exp. Pharmacol. Physiol. 2013, 40, 824–837.
- Hallfrisch, J. Metabolic effects of dietary fructose. FASEB J. 1990, 4, 2652–2660.
- Tappy, L.; Le, K.A. Metabolic effects of fructose and the worldwide increase in obesity. Physiol. Rev. 2010, 90, 23–46.
- Tappy, L.; Lê, K.A.; Tran, C.; Paquot, N. Fructose and metabolic diseases: New findings, new questions. Nutrition 2010, 26, 1044–1049.
- Campbell, E.; Schlappal, A.; Geller, E.; Castonguay, T.W. Fructose-Induced Hypertriglyceridemia: A Review. In Nutrition in the Prevention and Treatment of Abdominal Obesity; Elsevier: Amsterdam, The Netherlands, 2014; pp. 197–205.
- Elliott, S.S.; Keim, N.L.; Stern, J.S.; Teff, K.; Havel, P.J. Fructose, weight gain, and the insulin resistance syndrome. Am. J. Clin. Nutr. 2002, 76, 911–922.
- Khitan, Z.; Kim, D.H. Fructose: A key factor in the development of metabolic syndrome and hypertension. J. Nutr. Metab. 2013, 2013, 682673.
- Laughlin, M.R. Normal roles for dietary fructose in carbohydrate metabolism. Nutrients 2014, 6, 3117–3129.
- Berg, J.M.; Tymoczko, J.L.; Stryer, L. The Utilization of Fatty Acids as Fuel Requires Three Stages of Processing. In Biochemistry, 5th ed.; W. H. Freeman: New York, NY, USA, 2002; pp. 903–906.
- Havel, P.J. Dietary fructose: Implications for dysregulation of energy homeostasis and lipid/carbohydrate metabolism. Nutr. Rev. 2005, 63, 133–137.
- Basaranoglu, M.; Basaranoglu, G.; Sabuncu, T.; Sentürk, H. Fructose as a key player in the development of fatty liver disease. World J. Gastroenterol. 2013, 19, 1166–1172.
- Softic, S.; Cohen, D.E.; Kahn, C.R. Role of Dietary Fructose and Hepatic De Novo Lipogenesis in Fatty Liver Disease. Dig. Dis. Sci. 2016, 61, 1282–1293.
- Hengist, A.; Koumanov, F.; Gonzalez, J.T. Fructose and metabolic health: Governed by hepatic glycogen status? J. Physiol. 2019, 597, 3573–3585.
- Ameer, F.; Scandiuzzi, L.; Hasnain, S.; Kalbacher, H.; Zaidi, N. De novo lipogenesis in health and disease. Metabolism 2014, 63, 895–902.
- Sanders, F.W.B.; Griffin, J.L. De novo lipogenesis in the liver in health and disease: More than just a shunting yard for glucose. Biol. Rev. 2016, 91, 452–468.
- Basciano, H.; Federico, L.; Adeli, K. Fructose, insulin resistance, and metabolic dyslipidemia. Nutr. Metab. 2005, 2, 5.
- Postic, C.; Girard, J. Contribution of de novo fatty acid synthesis to hepatic steatosis and insulin resistance: Lessons from genetically engineered mice. J. Clin. Investig. 2008, 118, 829–838.
- Thiam, A.R.; Farese, R.V.; Walther, T.C. The biophysics and cell biology of lipid droplets. Nat. Rev. Mol. Cell Biol. 2013, 14, 775–786.
- Song, Z.; Xiaoli, A.M.; Yang, F. Regulation and metabolic significance of De Novo lipogenesis in adipose tissues. Nutrients 2018, 10, 1383.
- Kawano, Y.; Cohen, D.E. Mechanisms of hepatic triglyceride accumulation in non-alcoholic fatty liver disease. J. Gastroenterol. 2013, 48, 434–441.
- Byrne, C.D.; Olufad, R.; Bruce, K.D.; Cagampang, F.R.; Ahmed, M.H. Metabolic disturbances in non-alcoholic fatty liver disease. Clin. Sci. 2009, 116, 539–564.
- Lee, W.-C.; Wu, K.L.H.; Leu, S.; Tain, Y.-L. Translational insights on developmental origins of metabolic syndrome: Focus on fructose consumption. Biomed. J. 2018, 41, 96–101.
- Smith, E.V.L.; Dyson, R.M.; Berry, M.J.; Gray, C. Fructose Consumption During Pregnancy Influences Milk Lipid Composition and Offspring Lipid Profiles in Guinea Pigs. Front. Endocrinol. 2020, 11, 550.
- Saad, A.F.; Dickerson, J.; Kechichian, T.B.; Yin, H.; Gamble, P.; Salazar, A.; Patrikeev, I.; Motamedi, M.; Saade, G.R.; Costantine, M.M. High-fructose diet in pregnancy leads to fetal programming of hypertension, insulin resistance, and obesity in adult offspring. Am. J. Obstet. Gynecol. 2016, 215, 985–994.
- Storlien, L.H.; Oakes, N.D.; Pan, D.A.; Kusunoki, M.; Jenkins, A.B. Syndromes of insulin resistance in the rat: Inducement by diet and amelioration with benfluorex. Diabetes 1993, 42, 457–462.
- Herman, R.; Zakim, D.; Stifel, F.B. Effect of diet on lipid metabolism in experimental animals and man. Fed. Proc. 1970, 29, 1302–1307.
- Inoue, I.; Takahashi, K.; Katayama, S.; Harada, Y.; Negishi, K.; Itabashi, A.; Ishii, J. Effect of troglitazone (CS-045) and bezafibrate on glucose tolerance, Liver Glycogen synthase activity, and β-oxidation in fructose-fed rats. Metabolism 1995, 44, 1626–1630.
- Okazaki, M.; Zhang, H.; Yoshida, Y.; Ichino, K.; Nakayama, S.; Oguchi, K. Correlation between Plasma Fibrinogen and Serum Lipids in Rats with Hyperlipidemia Induced by Cholesterol Free-High Fructose or High Cholesterol Diet. J. Nutr. Sci. Vitaminol. 1994, 40, 479–489.
- Martinez, F.J.; Rizza, R.A.; Romero, J.C. High-fructose feeding elicits insulin resistance, hyperinsulinism, and hypertension in normal mongrel dogs. Hypertension 1994, 23, 456–463.
- Srinivasan, S.R.; Clevidence, B.A.; Pargaonkar, P.S.; Radhakrishnamurthy, B.; Berenson, G.S. Varied effects of dietary sucrose and cholesterol on serum lipids, lipoproteins and apolipoproteins in rhesus monkeys. Atherosclerosis 1979, 33, 301–314.
- Stanhope, K.L.; Schwarz, J.M.; Keim, N.L.; Griffen, S.C.; Bremer, A.A.; Graham, J.L.; Hatcher, B.; Cox, C.L.; Dyachenko, A.; Zhang, W.; et al. Consuming fructose-sweetened, not glucose-sweetened, beverages increases visceral adiposity and lipids and decreases insulin sensitivity in overweight/obese humans. J. Clin. Investig. 2009, 119, 1322–1334.
- Theytaz, F.; de Giorgi, S.; Hodson, L.; Stefanoni, N.; Rey, V.; Schneiter, P.; Giusti, V.; Tappy, L. Metabolic fate of fructose ingested with and without glucose in a mixed meal. Nutrients 2014, 6, 2632–2649.
- Müller-Wille, S.; Rheinberger, H.-J. A Cultural History of Heredity; University of Chicago Press: Chicago, IL, USA, 2012; pp. xiii–323.
- Cordain, L.; Eaton, S.B.; Sebastian, A.; Mann, N.; Lindeberg, S.; Watkins, B.A.; O’Keefe, J.H.; Brand-Miller, J. Origins and evolution of the Western diet: Health implications for the 21st century. Am. J. Clin. Nutr. 2005, 81, 341–354.
- Johnson, R.J.; Andrews, P.; Benner, S.A.; Oliver, W.; Theodore, E. Woodward award. The evolution of obesity: Insights from the mid-Miocene. Trans. Am. Clin. Climatol. Assoc. 2010, 121, 295–308.
- Bidwell, A.J. Chronic Fructose Ingestion as a Major Health Concern: Is a Sedentary Lifestyle Making It Worse? A Review. Nutrients 2017, 9, 549.
- Lustig, R.H.; Schmidt, L.A.; Brindis, C.D. The toxic truth about sugar. Nature 2012, 482, 27–29.
- Johnson, R.J.; Segal, M.S.; Sautin, Y.; Nakagawa, T.; Feig, D.I.; Kang, D.-H.; Gersch, M.S.; Benner, S.; Sánchez-Lozada, L.G. Potential role of sugar (fructose) in the epidemic of hypertension, obesity and the metabolic syndrome, diabetes, kidney disease, and cardiovascular disease1–3. Am. J. Clin. Nutr. 2007, 86, 899–906.
- Abdulla, M.H.; Sattar, M.A.; Johns, E.J. The Relation between Fructose-Induced Metabolic Syndrome and Altered Renal Haemodynamic and Excretory Function in the Rat. Int. J. Nephrol. 2011, 2011, 934659.
- Lê, K.-A.; Ith, M.; Kreis, R.; Faeh, D.; Bortolotti, M.; Tran, C.; Boesch, C.; Tappy, L. Fructose overconsumption causes dyslipidemia and ectopic lipid deposition in healthy subjects with and without a family history of type 2 diabetes. Am. J. Clin. Nutr. 2009, 89, 1760–1765.
- McDevitt, R.M.; Poppitt, S.D.; Murgatroyd, P.R.; Prentice, A.M. Macronutrient disposal during controlled overfeeding with glucose, fructose, sucrose, or fat in lean and obese women. Am. J. Clin. Nutr. 2000, 72, 369–377.
- Sloboda, D.M.; Li, M.; Patel, R.; Clayton, Z.E.; Yap, C.; Vickers, M.H. Early life exposure to fructose and offspring phenotype: Implications for long term metabolic homeostasis. J. Obes. 2014, 2014, 203474.
- Herman, M.A.; Samuel, V.T. The Sweet Path to Metabolic Demise: Fructose and Lipid Synthesis. Trends Endocrinol. Metab. 2016, 27, 719–730.
- Stanhope, K.L.; Medici, V.; A Bremer, A.; Lee, V.; Lam, H.D.; Nunez, M.V.; Chen, G.X.; Keim, N.L.; Havel, P.J. A dose-response study of consuming high-fructose corn syrup–sweetened beverages on lipid/lipoprotein risk factors for cardiovascular disease in young adults. Am. J. Clin. Nutr. 2015, 101, 1144–1154.
- Prinz, P. The role of dietary sugars in health: Molecular composition or just calories? Eur. J. Clin. Nutr. 2019, 73, 1216–1223.
- Johnson, R.K.; Appel, L.J.; Brands, M.; Howard, B.V.; Lefevre, M.; Lustig, R.H.; Sacks, F.; Steffen, L.M.; Wylie-Rosett, J.; on behalf of the American Heart Association Nutrition Committee of the Council on Nutrition, Physical Activity, and Metabolism and the Council on Epidemiology and Prevention. Dietary sugars intake and cardiovascular health a scientific statement from the american heart association. Circulation 2009, 120, 1011–1020.
- Winett, L.; Wallack, L.; Richardson, D.; Boone-Heinonen, J.; Messer, L. A Framework to Address Challenges in Communicating the Developmental Origins of Health and Disease. Curr. Environ. Health Rep. 2016, 3, 169–177.
- Bayol, S.A.; Simbi, B.H.; Fowkes, R.C.; Stickland, N.C. A maternal “junk food” diet in pregnancy and lactation promotes nonalcoholic Fatty liver disease in rat offspring. Endocrinology 2010, 151, 1451–1461.
- Rawana, S.; Clark, K.; Zhong, S.; Buison, A.; Chackunkal, S.; Jen, K.-L.C. Low Dose Fructose Ingestion during Gestation and Lactation Affects Carbohydrate Metabolism in Rat Dams and Their Offspring. J. Nutr. 1993, 123, 2158–2165.
- Vickers, M.H.; Clayton, Z.E.; Yap, C.; Sloboda, D.M. Maternal Fructose Intake during Pregnancy and Lactation Alters Placental Growth and Leads to Sex-Specific Changes in Fetal and Neonatal Endocrine Function. Endocrinology 2011, 152, 1378–1387.
- Gray, C.; Gardiner, S.M.; Elmes, M.; Gardner, D.S. Excess maternal salt or fructose intake programmes sex-specific, stress-and fructose-sensitive hypertension in the offspring. Br. J. Nutr. 2016, 115, 594–604.
- Smith, E.V.L.; Dyson, R.M.; Vanderboor, C.M.G.; Sarr, O.; Anderson, J.; Berry, M.J.; Regnault, T.R.H.; Peng, L.; Gray, C. Maternal Fructose Intake Causes Developmental Reprogramming of Hepatic Mitochondrial Catalytic Activity and Lipid Metabolism in Weanling and Young Adult Offspring. Int. J. Mol. Sci. 2022, 23, 999.
- Smith, E. Excess Maternal Fructose Intake and the Developmental Programming of Mitochondrial Function and Lipid Metabolism in Adult Offspring. Ph.D. Thesis, University of Otago, Wellington, New Zealand, 2021.
- Goran, M.I.; Martin, A.A.; Alderete, T.L.; Fujiwara, H.; Fields, D.A. Fructose in breast milk is positively associated with infant body composition at 6 months of age. Nutrients 2017, 9, 146.
- Berger, P.K.; Fields, D.A.; Demerath, E.W.; Fujiwara, H.; Goran, M.I. High-fructose corn-syrup-sweetened beverage intake increases 5-hour breast milk fructose concentrations in lactating women. Nutrients 2018, 10, 669.
- Chappel, J.E.; Clandinin, M.T.; Kearney-Volpe, C. Trans fatty acids in human milk lipids: Influence of maternal diet and weight loss. Am. J. Clin. Nutr. 1985, 42, 49–56.
- Mennitti, L.V.; Oliveira, J.L.; Morais, C.A.; Estadella, D.; Oyama, L.M.; Oller do Nascimento, C.M.; Pisani, L.P. Type of fatty acids in maternal diets during pregnancy and/or lactation and metabolic consequences of the offspring. J. Nutr. Biochem. 2015, 26, 99–111.
- Carta, G.; Murru, E.; Banni, S.; Manca, C. Palmitic acid: Physiological role, metabolism and nutritional implications. Front. Physiol. 2017, 8, 902.
- Ogawa, Y.; Imajo, K.; Honda, Y.; Kessoku, T.; Tomeno, W.; Kato, S.; Fujita, K.; Yoneda, M.; Saito, S.; Saigusa, Y.; et al. Palmitate-induced lipotoxicity is crucial for the pathogenesis of nonalcoholic fatty liver disease in cooperation with gut-derived endotoxin. Sci. Rep. 2018, 8, 11365.
- Okada, T.; Furuhashi, N.; Kuromori, Y.; Miyashita, M.; Iwata, F.; Harada, K. Plasma palmitoleic acid content and obesity in children. Am. J. Clin. Nutr. 2005, 82, 747–750.
- Hannou, S.A.; Haslam, D.E.; McKeown, N.M.; Herman, M.A. Fructose metabolism and metabolic disease. J. Clin. Investig. 2018, 128, 545–555.
- Bezerra, R.; Ueno, M.; Silva, M.; Tavares, D.; Carvalho, C.; Saad, M.; Gontijo, J. A high-fructose diet induces insulin resistance but not blood pressure changes in normotensive rats. Braz. J. Med. Biol. Res. 2001, 34, 1155–1160.
- Hawkins, M.; Gabriely, I.; Wozniak, R.; Vilcu, C.; Shamoon, H.; Rossetti, L. Fructose improves the ability of hyperglycemia per se to regulate glucose production in type 2 diabetes. Diabetes 2002, 51, 606–614.
- Abraha, A.; Humphreys, S.M.; Clark, M.L.; Matthews, D.R.; Frayn, K.N. Acute effect of fructose on postprandial lipaemia in diabetic and non-diabetic subjects. Br. J. Nutr. 1998, 80, 169–175.
- Patel, C.; Douard, V.; Yu, S.; Gao, N.; Ferraris, R.P. Transport, metabolism, and endosomal trafficking-dependent regulation of intestinal fructose absorption. FASEB J. 2015, 29, 4046–4058.
- Jornayvaz, F.R.; Shulman, G.I. Diacylglycerol activation of protein kinase Cε and hepatic insulin resistance. Cell Metab. 2012, 15, 574–584.
- Samuel, V.T.; Shulman, G.I. Mechanisms for insulin resistance: Common threads and missing links. Cell 2012, 148, 852–871.
- Myers, M.G.; Cowley, M.A.; Münzberg, H. Mechanisms of leptin action and leptin resistance. Annu. Rev. Physiol. 2008, 70, 537–556.
- Shapiro, A.; Mu, W.; Roncal, C.; Cheng, K.Y.; Johnson, R.J.; Scarpace, P.J. Fructose-induced leptin resistance exacerbates weight gain in response to subsequent high-fat feeding. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2008, 295, 1370–1375.
- Cummings, D.E.; Shannon, M.H. Roles for ghrelin in the regulation of appetite and body weight. In Archives of Surgery; American Medical Association: Seattle, WA, USA, 2003; pp. 389–396.
- Kojima, M.; Kangawa, K. Ghrelin: Structure and function. Am. Physiol. Soc. 2005, 85, 495–522.
- Ma, X.; Lin, L.; Yue, J.; Wu, C.-S.; Guo, C.A.; Wang, R.; Yu, K.-J.; Devaraj, S.; Murano, P.; Chen, Z.; et al. Suppression of Ghrelin Exacerbates HFCS-Induced Adiposity and Insulin Resistance. Int. J. Mol. Sci. 2017, 18, 1302.
- Teff, K.L.; Elliott, S.S.; Tschöp, M.; Kieffer, T.J.; Rader, D.; Heiman, M.; Townsend, R.R.; Keim, N.L.; D’Alessio, D.; Havel, P.J.; et al. Dietary Fructose Reduces Circulating Insulin and Leptin, Attenuates Postprandial Suppression of Ghrelin, and Increases Triglycerides in Women. J. Clin. Endocrinol. Metab. 2004, 89, 2963–2972.
- Kisioglu, B.; Nergiz-Unal, R. Potential effect of maternal dietary sucrose or fructose syrup on CD36, leptin, and ghrelin-mediated fetal programming of obesity. Nutr. Neurosci. 2020, 23, 210–220.
- Rodríguez, L.; Panadero, M.I.; Roglans, N.; Otero, P.; Álvarez-Millán, J.J.; Laguna, J.C.; Bocos, C. Fructose during pregnancy affects maternal and fetal leptin signaling. J. Nutr. Biochem. 2013, 24, 1709–1716.
- Jen, K.L.C.; Rochon, C.; Zhong, S.; Whitcomb, L. Fructose and sucrose feeding during pregnancy and lactation in rats changes maternal and pup fuel metabolism. J. Nutr. 1991, 121, 1999–2005.


