 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Linlin Guo | -- | 3841 | 2022-10-21 06:20:10 | | | |
| 2 | Lindsay Dong | -2 word(s) | 3839 | 2022-10-21 07:59:16 | | | | |
| 3 | Lindsay Dong | Meta information modification | 3839 | 2022-10-26 07:52:44 | | |
Video Upload Options
The multifunctional protein, voltage-dependent anion channel 1 (VDAC1), is located on the mitochondrial outer membrane. It is a pivotal protein that maintains mitochondrial function to power cellular bioactivities via energy generation. VDAC1 is involved in regulating energy production, mitochondrial oxidase stress, Ca2+ transportation, substance metabolism, apoptosis, mitochondrial autophagy (mitophagy), and many other functions. VDAC1 malfunction is associated with mitochondrial disorders that affect inflammatory responses, resulting in an up-regulation of the body’s defensive response to stress stimulation. Overresponses to inflammation may cause chronic diseases. Mitochondrial DNA (mtDNA) acts as a danger signal that can further trigger native immune system activities after its secretion. VDAC1 mediates the release of mtDNA into the cytoplasm to enhance cytokine levels by activating immune responses. VDAC1 regulates mitochondrial Ca2+ transportation, lipid metabolism and mitophagy, which are involved in inflammation-related disease pathogenesis.
1. Introduction
2. Inflammation, VDAC1 Mediates Apoptosis and Mitochondrial Oxidative Stress
2.1. VDAC1 Regulates Inflammation via Mediating Apoptosis
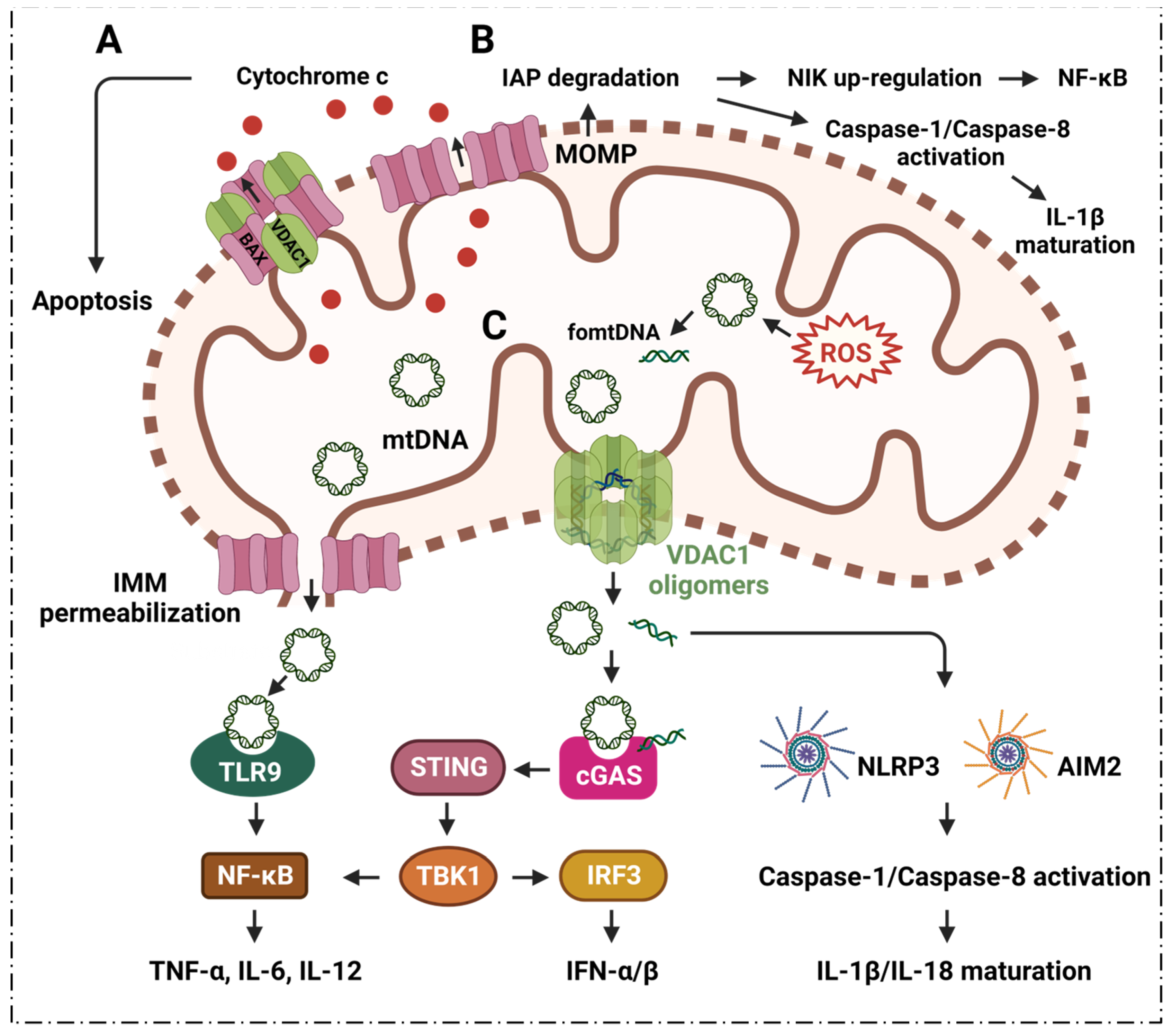
2.2. VDAC1 Mediates Mitochondrial Oxidative Stress in Immune Responses
3. Inflammation and VDAC1 Mediates Mitochondrial Ca2+ Transportation
3.1. Neutrophils
3.2. Macrophages
3.3. Dendritic Cells
4. Inflammatory Diseases and VDAC1 in Energy Metabolism
4.1. TCA Cycle
4.2. Glycolysis
5. Inflammatory Diseases and VDAC1 in Lipid Metabolism
6. Inflammatory Diseases Pathogenesis and VDAC1 in Mitophagy
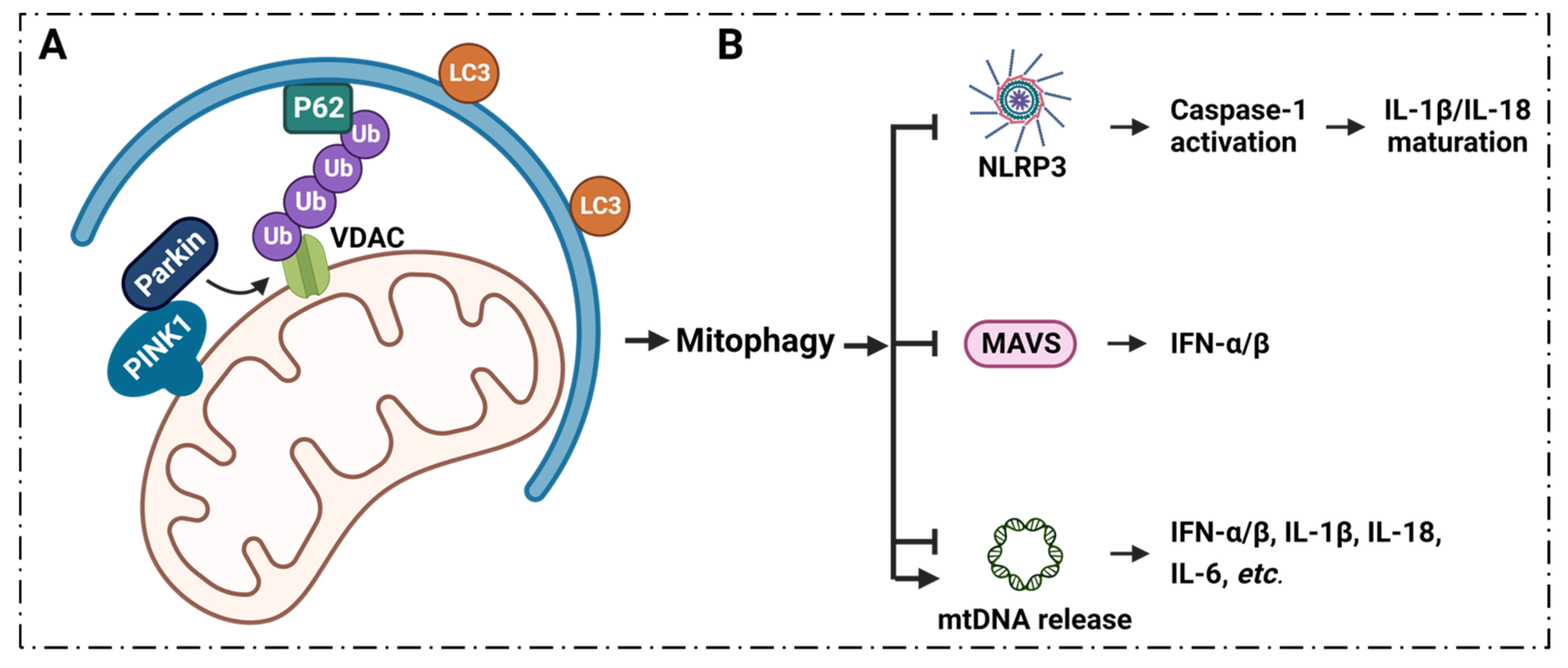
6.1. Mitophagy Regulates Inflammation via VDAC1
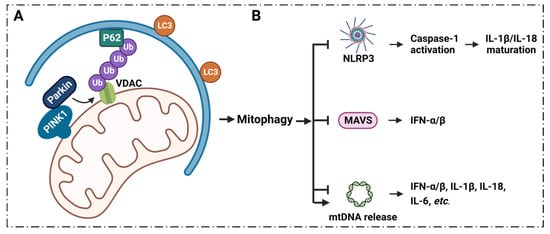
7. Summary and Conclusions, Current Clinical Conditions and Future Perspectives
References
- Medzhitov, R. Inflammation 2010: New adventures of an old flame. Cell 2010, 140, 771–776.
- Scrivo, R.; Vasile, M.; Bartosiewicz, I.; Valesini, G. Inflammation as “common soil” of the multifactorial diseases. Autoimmun. Rev. 2011, 10, 369–374.
- Medzhitov, R. Origin and physiological roles of inflammation. Nature 2008, 454, 428–435.
- Hengartner, M.O. The biochemistry of apoptosis. Nature 2000, 407, 770–776.
- Nunnari, J.; Suomalainen, A. Mitochondria: In sickness and in health. Cell 2012, 148, 1145–1159.
- Pfanner, N.; Warscheid, B.; Wiedemann, N. Mitochondrial proteins: From biogenesis to functional networks. Nat. Rev. Mol. Cell Biol. 2019, 20, 267–284.
- Bock, F.J.; Tait, S.W.G. Mitochondria as multifaceted regulators of cell death. Nat. Rev. Mol. Cell Biol. 2020, 21, 85–100.
- Tait, S.W.; Green, D.R. Mitochondrial regulation of cell death. Cold Spring Harb. Perspect. Biol. 2013, 5, a008706.
- Marchi, S.; Guilbaud, E.; Tait, S.W.G.; Yamazaki, T.; Galluzzi, L. Mitochondrial control of inflammation. Nat. Rev. Immunol. 2022, 1–15.
- Schapira, A.H. Mitochondrial diseases. Lancet 2012, 379, 1825–1834.
- Gorman, G.S.; Chinnery, P.F.; DiMauro, S.; Hirano, M.; Koga, Y.; McFarland, R.; Suomalainen, A.; Thorburn, D.R.; Zeviani, M.; Turnbull, D.M. Mitochondrial diseases. Nat. Rev. Dis. Prim. 2016, 2, 16080.
- Wallace, D.C. Mitochondrial genetic medicine. Nat. Genet. 2018, 50, 1642–1649.
- Schein, S.J.; Colombini, M.; Finkelstein, A. Reconstitution in planar lipid bilayers of a voltage-dependent anion-selective channel obtained from paramecium mitochondria. J. Membr. Biol. 1976, 30, 99–120.
- Di Rosa, M.C.; Guarino, F.; Conti Nibali, S.; Magrì, A.; De Pinto, V. Voltage-Dependent Anion Selective Channel Isoforms in Yeast: Expression, Structure, and Functions. Front. Physiol. 2021, 12, 675708.
- Guardiani, C.; Magrì, A.; Karachitos, A.; Di Rosa, M.C.; Reina, S.; Bodrenko, I.; Messina, A.; Kmita, H.; Ceccarelli, M.; De Pinto, V. yVDAC2, the second mitochondrial porin isoform of Saccharomyces cerevisiae. Biochim. Biophys. Acta Bioenerg. 2018, 1859, 270–279.
- De Stefani, D.; Bononi, A.; Romagnoli, A.; Messina, A.; De Pinto, V.; Pinton, P.; Rizzuto, R. VDAC1 selectively transfers apoptotic Ca2+ signals to mitochondria. Cell Death Differ. 2012, 19, 267–273.
- Messina, A.; Reina, S.; Guarino, F.; De Pinto, V. VDAC isoforms in mammals. Biochim. Biophys. Acta 2012, 1818, 1466–1476.
- Zinghirino, F.; Pappalardo, X.G.; Messina, A.; Guarino, F.; De Pinto, V. Is the secret of VDAC Isoforms in their gene regulation? Characterization of human VDAC genes expression profile, promoter activity, and transcriptional regulators. Int. J. Mol. Sci. 2020, 21, 7388.
- Cheng, E.H.; Sheiko, T.V.; Fisher, J.K.; Craigen, W.J.; Korsmeyer, S.J. VDAC2 inhibits BAK activation and mitochondrial apoptosis. Science 2003, 301, 513–517.
- Reina, S.; Nibali, S.C.; Tomasello, M.F.; Magrì, A.; Messina, A.; De Pinto, V. Voltage Dependent Anion Channel 3 (VDAC3) protects mitochondria from oxidative stress. Redox Biol. 2022, 51, 102264.
- Zinghirino, F.; Pappalardo, X.G.; Messina, A.; Nicosia, G.; De Pinto, V.; Guarino, F. VDAC Genes Expression and Regulation in Mammals. Front. Physiol. 2021, 12, 708695.
- Hiller, S.; Garces, R.G.; Malia, T.J.; Orekhov, V.Y.; Colombini, M.; Wagner, G. Solution structure of the integral human membrane protein VDAC-1 in detergent micelles. Science 2008, 321, 1206–1210.
- Ham, S.J.; Lee, D.; Yoo, H.; Jun, K.; Shin, H.; Chung, J. Decision between mitophagy and apoptosis by Parkin via VDAC1 ubiquitination. Proc. Natl. Acad. Sci. USA 2020, 117, 4281–4291.
- Shoshan-Barmatz, V.; Golan, M. Mitochondrial VDAC1: Function in cell life and death and a target for cancer therapy. Curr. Med. Chem. 2012, 19, 714–735.
- Shoshan-Barmatz, V.; De Pinto, V.; Zweckstetter, M.; Raviv, Z.; Keinan, N.; Arbel, N. VDAC, a multi-functional mitochondrial protein regulating cell life and death. Mol. Asp. Med. 2010, 31, 227–285.
- Huang, H.; Hu, X.; Eno, C.O.; Zhao, G.; Li, C.; White, C. An interaction between Bcl-xL and the voltage-dependent anion channel (VDAC) promotes mitochondrial Ca2+ uptake. J. Biol. Chem. 2013, 288, 19870–19881.
- Baines, C.P.; Kaiser, R.A.; Sheiko, T.; Craigen, W.J.; Molkentin, J.D. Voltage-dependent anion channels are dispensable for mitochondrial-dependent cell death. Nat. Cell Biol. 2007, 9, 550–555.
- Kim, J.; Gupta, R.; Blanco, L.P.; Yang, S.; Shteinfer-Kuzmine, A.; Wang, K.; Zhu, J.; Yoon, H.E.; Wang, X.; Kerkhofs, M.; et al. VDAC oligomers form mitochondrial pores to release mtDNA fragments and promote lupus-like disease. Science 2019, 366, 1531–1536.
- Xian, H.; Watari, K.; Sanchez-Lopez, E.; Offenberger, J.; Onyuru, J.; Sampath, H.; Ying, W.; Hoffman, H.M.; Shadel, G.S.; Karin, M. Oxidized DNA fragments exit mitochondria via mPTP- and VDAC-dependent channels to activate NLRP3 inflammasome and interferon signaling. Immunity 2022, 55, 1370–1385.e1378.
- Wu, J.; Ma, Z.; Raman, A.; Beckerman, P.; Dhillon, P.; Mukhi, D.; Palmer, M.; Chen, H.C.; Cohen, C.R.; Dunn, T.; et al. APOL1 risk variants in individuals of African genetic ancestry drive endothelial cell defects that exacerbate sepsis. Immunity 2021, 54, 2632–2649.e2636.
- Onishi, M.; Yamano, K.; Sato, M.; Matsuda, N.; Okamoto, K. Molecular mechanisms and physiological functions of mitophagy. EMBO J. 2021, 40, e104705.
- Marek-Iannucci, S.; Ozdemir, A.B.; Moreira, D.; Gomez, A.C.; Lane, M.; Porritt, R.A.; Lee, Y.; Shimada, K.; Abe, M.; Stotland, A.; et al. Autophagy-mitophagy induction attenuates cardiovascular inflammation in a murine model of Kawasaki disease vasculitis. JCI Insight 2021, 6, e151981.
- Green, D.R.; Galluzzi, L.; Kroemer, G. Mitochondria and the autophagy-inflammation-cell death axis in organismal aging. Science 2011, 333, 1109–1112.
- Sidarala, V.; Pearson, G.L.; Parekh, V.S.; Thompson, B.; Christen, L.; Gingerich, M.A.; Zhu, J.; Stromer, T.; Ren, J.; Reck, E.C.; et al. Mitophagy protects β cells from inflammatory damage in diabetes. JCI Insight 2020, 5, e141138.
- Ip, W.K.E.; Hoshi, N.; Shouval, D.S.; Snapper, S.; Medzhitov, R. Anti-inflammatory effect of IL-10 mediated by metabolic reprogramming of macrophages. Science 2017, 356, 513–519.
- Camara, A.K.S.; Zhou, Y.; Wen, P.C.; Tajkhorshid, E.; Kwok, W.M. Mitochondrial VDAC1: A Key Gatekeeper as Potential Therapeutic Target. Front. Physiol. 2017, 8, 460.
- Suomalainen, A.; Battersby, B.J. Mitochondrial diseases: The contribution of organelle stress responses to pathology. Nat. Rev. Mol. Cell Biol. 2018, 19, 77–92.
- Giorgio, V.; Guo, L.; Bassot, C.; Petronilli, V.; Bernardi, P. Calcium and regulation of the mitochondrial permeability transition. Cell Calcium 2018, 70, 56–63.
- Urbani, A.; Giorgio, V.; Carrer, A.; Franchin, C.; Arrigoni, G.; Jiko, C.; Abe, K.; Maeda, S.; Shinzawa-Itoh, K.; Bogers, J.F.M.; et al. Purified F-ATP synthase forms a Ca2+-dependent high-conductance channel matching the mitochondrial permeability transition pore. Nat. Commun. 2019, 10, 4341.
- Moya, G.E.; Rivera, P.D.; Dittenhafer-Reed, K.E. Evidence for the Role of Mitochondrial DNA Release in the Inflammatory Response in Neurological Disorders. Int. J. Mol. Sci. 2021, 22, 7030.
- Gutiérrez-Aguilar, M.; Baines, C.P. Structural mechanisms of cyclophilin D-dependent control of the mitochondrial permeability transition pore. Biochim. Biophys. Acta 2015, 1850, 2041–2047.
- Jia, K.; Du, H. Mitochondrial Permeability Transition: A Pore Intertwines Brain Aging and Alzheimer’s Disease. Cells 2021, 10, 649.
- Green, D.R.; Reed, J.C. Mitochondria and apoptosis. Science 1998, 281, 1309–1312.
- Shimizu, S.; Narita, M.; Tsujimoto, Y. Bcl-2 family proteins regulate the release of apoptogenic cytochrome c by the mitochondrial channel VDAC. Nature 1999, 399, 483–487.
- Yang, J.; Liu, X.; Bhalla, K.; Kim, C.N.; Ibrado, A.M.; Cai, J.; Peng, T.I.; Jones, D.P.; Wang, X. Prevention of apoptosis by Bcl-2: Release of cytochrome c from mitochondria blocked. Science 1997, 275, 1129–1132.
- Peña-Blanco, A.; García-Sáez, A.J. Bax, Bak and beyond—Mitochondrial performance in apoptosis. FEBS J. 2018, 285, 416–431.
- Banerjee, J.; Ghosh, S. Bax increases the pore size of rat brain mitochondrial voltage-dependent anion channel in the presence of tBid. Biochem. Biophys. Res. Commun. 2004, 323, 310–314.
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516.
- Wang, C.; Youle, R.J. The role of mitochondria in apoptosis. Annu. Rev. Genet. 2009, 43, 95–118.
- Pérez-Treviño, P.; Velásquez, M.; García, N. Mechanisms of mitochondrial DNA escape and its relationship with different metabolic diseases. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165761.
- Alexeyev, M.; Shokolenko, I.; Wilson, G.; LeDoux, S. The maintenance of mitochondrial DNA integrity—critical analysis and update. Cold Spring Harb. Perspect. Biol. 2013, 5, a012641.
- Moreira, P.I.; Carvalho, C.; Zhu, X.; Smith, M.A.; Perry, G. Mitochondrial dysfunction is a trigger of Alzheimer’s disease pathophysiology. Biochim. Biophys. Acta 2010, 1802, 2–10.
- Schmitt, K.; Grimm, A.; Kazmierczak, A.; Strosznajder, J.B.; Götz, J.; Eckert, A. Insights into mitochondrial dysfunction: Aging, amyloid-β, and tau-A deleterious trio. Antioxid. Redox Signal. 2012, 16, 1456–1466.
- Lee, S.R.; Han, J. Mitochondrial Nucleoid: Shield and Switch of the Mitochondrial Genome. Oxidative Med. Cell. Longev. 2017, 2017, 8060949.
- Ben-Hail, D.; Begas-Shvartz, R.; Shalev, M.; Shteinfer-Kuzmine, A.; Gruzman, A.; Reina, S.; De Pinto, V.; Shoshan-Barmatz, V. Novel Compounds Targeting the Mitochondrial Protein VDAC1 Inhibit Apoptosis and Protect against Mitochondrial Dysfunction. J. Biol. Chem. 2016, 291, 24986–25003.
- Shteinfer-Kuzmine, A.; Argueti-Ostrovsky, S.; Leyton-Jaimes, M.F.; Anand, U.; Abu-Hamad, S.; Zalk, R.; Shoshan-Barmatz, V.; Israelson, A. Targeting the Mitochondrial Protein VDAC1 as a Potential Therapeutic Strategy in ALS. Int. J. Mol. Sci. 2022, 23, 9946.
- Zhang, E.; Mohammed Al-Amily, I.; Mohammed, S.; Luan, C.; Asplund, O.; Ahmed, M.; Ye, Y.; Ben-Hail, D.; Soni, A.; Vishnu, N.; et al. Preserving Insulin Secretion in Diabetes by Inhibiting VDAC1 Overexpression and Surface Translocation in β Cells. Cell Metab. 2019, 29, 64–77.e66.
- Klapper-Goldstein, H.; Verma, A.; Elyagon, S.; Gillis, R.; Murninkas, M.; Pittala, S.; Paul, A.; Shoshan-Barmatz, V.; Etzion, Y. VDAC1 in the diseased myocardium and the effect of VDAC1-interacting compound on atrial fibrosis induced by hyperaldosteronism. Sci. Rep. 2020, 10, 22101.
- Verma, A.; Pittala, S.; Alhozeel, B.; Shteinfer-Kuzmine, A.; Ohana, E.; Gupta, R.; Chung, J.H.; Shoshan-Barmatz, V. The role of the mitochondrial protein VDAC1 in inflammatory bowel disease: A potential therapeutic target. Mol. Ther. J. Am. Soc. Gene Ther. 2022, 30, 726–744.
- Bravo-Sagua, R.; Parra, V.; López-Crisosto, C.; Díaz, P.; Quest, A.F.; Lavandero, S. Calcium Transport and Signaling in Mitochondria. Compr. Physiol. 2017, 7, 623–634.
- Shoshan-Barmatz, V.; De, S. Mitochondrial VDAC, the Na+/Ca2+ Exchanger, and the Ca2+ Uniporter in Ca2+ Dynamics and Signaling. Adv. Exp. Med. Biol. 2017, 981, 323–347.
- Ben-Hail, D.; Palty, R.; Shoshan-Barmatz, V. Measurement of mitochondrial Ca2+ transport mediated by three transport proteins: VDAC1, the Na+/Ca2+ exchanger, and the Ca2+ uniporter. Cold Spring Harb. Protoc. 2014, 2014, 161–166.
- Gincel, D.; Zaid, H.; Shoshan-Barmatz, V. Calcium binding and translocation by the voltage-dependent anion channel: A possible regulatory mechanism in mitochondrial function. Biochem. J. 2001, 358, 147–155.
- Abu-Hamad, S.; Sivan, S.; Shoshan-Barmatz, V. The expression level of the voltage-dependent anion channel controls life and death of the cell. Proc. Natl. Acad. Sci. USA 2006, 103, 5787–5792.
- Griesbeck, O.; Baird, G.S.; Campbell, R.E.; Zacharias, D.A.; Tsien, R.Y. Reducing the environmental sensitivity of yellow fluorescent protein. Mechanism and applications. J. Biol. Chem. 2001, 276, 29188–29194.
- Dalal, P.J.; Muller, W.A.; Sullivan, D.P. Endothelial Cell Calcium Signaling during Barrier Function and Inflammation. Am. J. Pathol. 2020, 190, 535–542.
- Berridge, M.J.; Bootman, M.D.; Lipp, P. Calcium—A life and death signal. Nature 1998, 395, 645–648.
- Lokuta, M.A.; Nuzzi, P.A.; Huttenlocher, A. Calpain regulates neutrophil chemotaxis. Proc. Natl. Acad. Sci. USA 2003, 100, 4006–4011.
- Kolaczkowska, E.; Kubes, P. Neutrophil recruitment and function in health and inflammation. Nat. Rev. Immunol. 2013, 13, 159–175.
- Immler, R.; Simon, S.I.; Sperandio, M. Calcium signalling and related ion channels in neutrophil recruitment and function. Eur. J. Clin. Investig. 2018, 48 (Suppl. S2), e12964.
- Dewitt, S.; Francis, R.J.; Hallett, M.B. Ca²⁺ and calpain control membrane expansion during the rapid cell spreading of neutrophils. J. Cell Sci. 2013, 126 Pt 20, 4627–4635.
- Nguyen, G.T.; Green, E.R.; Mecsas, J. Neutrophils to the ROScue: Mechanisms of NADPH Oxidase Activation and Bacterial Resistance. Front. Cell. Infect. Microbiol. 2017, 7, 373.
- Bouti, P.; Webbers, S.D.S.; Fagerholm, S.C.; Alon, R.; Moser, M.; Matlung, H.L.; Kuijpers, T.W. β2 Integrin Signaling Cascade in Neutrophils: More Than a Single Function. Front. Immunol. 2021, 11, 619925.
- Chen, H.; Gao, W.; Yang, Y.; Guo, S.; Wang, H.; Wang, W.; Zhang, S.; Zhou, Q.; Xu, H.; Yao, J.; et al. Inhibition of VDAC1 prevents Ca²⁺-mediated oxidative stress and apoptosis induced by 5-aminolevulinic acid mediated sonodynamic therapy in THP-1 macrophages. Apoptosis Int. J. Program. Cell Death 2014, 19, 1712–1726.
- Danelishvili, L.; Chinison, J.J.J.; Pham, T.; Gupta, R.; Bermudez, L.E. The Voltage-Dependent Anion Channels (VDAC) of Mycobacterium avium phagosome are associated with bacterial survival and lipid export in macrophages. Sci. Rep. 2017, 7, 7007.
- Hirayama, D.; Iida, T.; Nakase, H. The Phagocytic Function of Macrophage-Enforcing Innate Immunity and Tissue Homeostasis. Int. J. Mol. Sci. 2017, 19, 92.
- Arango Duque, G.; Descoteaux, A. Macrophage cytokines: Involvement in immunity and infectious diseases. Front. Immunol. 2014, 5, 491.
- Nunes, P.; Demaurex, N. The role of calcium signaling in phagocytosis. J. Leukoc. Biol. 2010, 88, 57–68.
- Tedesco, S.; Scattolini, V.; Albiero, M.; Bortolozzi, M.; Avogaro, A.; Cignarella, A.; Fadini, G.P. Mitochondrial Calcium Uptake Is Instrumental to Alternative Macrophage Polarization and Phagocytic Activity. Int. J. Mol. Sci. 2019, 20, 4966.
- Paredes-Gamero, E.J.; Leon, C.M.; Borojevic, R.; Oshiro, M.E.; Ferreira, A.T. Changes in intracellular Ca2+ levels induced by cytokines and P2 agonists differentially modulate proliferation or commitment with macrophage differentiation in murine hematopoietic cells. J. Biol. Chem. 2008, 283, 31909–31919.
- Kang, H.; Zhang, K.; Wong, D.S.H.; Han, F.; Li, B.; Bian, L. Near-infrared light-controlled regulation of intracellular calcium to modulate macrophage polarization. Biomaterials 2018, 178, 681–696.
- Hilligan, K.L.; Ronchese, F. Antigen presentation by dendritic cells and their instruction of CD4+ T helper cell responses. Cell. Mol. Immunol. 2020, 17, 587–599.
- Sumoza-Toledo, A.; Lange, I.; Cortado, H.; Bhagat, H.; Mori, Y.; Fleig, A.; Penner, R.; Partida-Sánchez, S. Dendritic cell maturation and chemotaxis is regulated by TRPM2-mediated lysosomal Ca2+ release. FASEB J. 2011, 25, 3529–3542.
- Worbs, T.; Hammerschmidt, S.I.; Förster, R. Dendritic cell migration in health and disease. Nat. Rev. Immunol. 2017, 17, 30–48.
- Braun, A.; Worbs, T.; Moschovakis, G.L.; Halle, S.; Hoffmann, K.; Bölter, J.; Münk, A.; Förster, R. Afferent lymph-derived T cells and DCs use different chemokine receptor CCR7-dependent routes for entry into the lymph node and intranodal migration. Nat. Immunol. 2011, 12, 879–887.
- Shao, Z.; Makinde, T.O.; Agrawal, D.K. Calcium-activated potassium channel KCa3.1 in lung dendritic cell migration. Am. J. Respir. Cell Mol. Biol. 2011, 45, 962–968.
- Rodríguez-Fernández, J.L.; Criado-García, O. The Chemokine Receptor CCR7 Uses Distinct Signaling Modules With Biased Functionality to Regulate Dendritic Cells. Front. Immunol. 2020, 11, 528.
- Riol-Blanco, L.; Sánchez-Sánchez, N.; Torres, A.; Tejedor, A.; Narumiya, S.; Corbí, A.L.; Sánchez-Mateos, P.; Rodríguez-Fernández, J.L. The chemokine receptor CCR7 activates in dendritic cells two signaling modules that independently regulate chemotaxis and migratory speed. J. Immunol. 2005, 174, 4070–4080.
- Viola, A.; Munari, F.; Sánchez-Rodríguez, R.; Scolaro, T.; Castegna, A. The Metabolic Signature of Macrophage Responses. Front. Immunol. 2019, 10, 1462.
- Tannahill, G.M.; Curtis, A.M.; Adamik, J.; Palsson-McDermott, E.M.; McGettrick, A.F.; Goel, G.; Frezza, C.; Bernard, N.J.; Kelly, B.; Foley, N.H.; et al. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature 2013, 496, 238–242.
- Littlewood-Evans, A.; Sarret, S.; Apfel, V.; Loesle, P.; Dawson, J.; Zhang, J.; Muller, A.; Tigani, B.; Kneuer, R.; Patel, S.; et al. GPR91 senses extracellular succinate released from inflammatory macrophages and exacerbates rheumatoid arthritis. J. Exp. Med. 2016, 213, 1655–1662.
- Mills, E.; O’Neill, L.A. Succinate: A metabolic signal in inflammation. Trends Cell Biol. 2014, 24, 313–320.
- Roberts, D.J.; Miyamoto, S. Hexokinase II integrates energy metabolism and cellular protection: Akting on mitochondria and TORCing to autophagy. Cell Death Differ. 2015, 22, 248–257.
- Zhang, D.; Yip, Y.M.; Li, L. In silico construction of HK2-VDAC1 complex and investigating the HK2 binding-induced molecular gating mechanism of VDAC1. Mitochondrion 2016, 30, 222–228.
- Xu, D.; Jin, J.; Yu, H.; Zhao, Z.; Ma, D.; Zhang, C.; Jiang, H. Chrysin inhibited tumor glycolysis and induced apoptosis in hepatocellular carcinoma by targeting hexokinase-2. J. Exp. Clin. Cancer Res. CR 2017, 36, 44.
- Mills, E.L.; Kelly, B.; O’Neill, L.A.J. Mitochondria are the powerhouses of immunity. Nat. Immunol. 2017, 18, 488–498.
- Shoshan-Barmatz, V.; Ben-Hail, D.; Admoni, L.; Krelin, Y.; Tripathi, S.S. The mitochondrial voltage-dependent anion channel 1 in tumor cells. Biochim. Biophys. Acta 2015, 1848, 2547–2575.
- Shoshan-Barmatz, V.; Pittala, S.; Mizrachi, D. VDAC1 and the TSPO: Expression, Interactions, and Associated Functions in Health and Disease States. Int. J. Mol. Sci. 2019, 20, 3348.
- Lee, K.; Kerner, J.; Hoppel, C.L. Mitochondrial carnitine palmitoyltransferase 1a (CPT1a) is part of an outer membrane fatty acid transfer complex. J. Biol. Chem. 2011, 286, 25655–25662.
- Martel, C.; Allouche, M.; Esposti, D.D.; Fanelli, E.; Boursier, C.; Henry, C.; Chopineau, J.; Calamita, G.; Kroemer, G.; Lemoine, A.; et al. Glycogen synthase kinase 3-mediated voltage-dependent anion channel phosphorylation controls outer mitochondrial membrane permeability during lipid accumulation. Hepatology 2013, 57, 93–102.
- Pittala, S.; Krelin, Y.; Kuperman, Y.; Shoshan-Barmatz, V. A Mitochondrial VDAC1-Based Peptide Greatly Suppresses Steatosis and NASH-Associated Pathologies in a Mouse Model. Mol. Ther. J. Am. Soc. Gene Ther. 2019, 27, 1848–1862.
- Turkaly, P.; Kerner, J.; Hoppel, C. A 22 kDa polyanion inhibits carnitine-dependent fatty acid oxidation in rat liver mitochondria. FEBS Lett. 1999, 460, 241–245.
- Xu, Y.; Shen, J.; Ran, Z. Emerging views of mitophagy in immunity and autoimmune diseases. Autophagy 2020, 16, 3–17.
- Ziegler, P.K.; Bollrath, J.; Pallangyo, C.K.; Matsutani, T.; Canli, Ö.; De Oliveira, T.; Diamanti, M.A.; Müller, N.; Gamrekelashvili, J.; Putoczki, T.; et al. Mitophagy in Intestinal Epithelial Cells Triggers Adaptive Immunity during Tumorigenesis. Cell 2018, 174, 88–101.e116.
- Ma, K.; Chen, G.; Li, W.; Kepp, O.; Zhu, Y.; Chen, Q. Mitophagy, Mitochondrial Homeostasis, and Cell Fate. Front. Cell Dev. Biol. 2020, 8, 467.
- Matsuda, N.; Sato, S.; Shiba, K.; Okatsu, K.; Saisho, K.; Gautier, C.A.; Sou, Y.S.; Saiki, S.; Kawajiri, S.; Sato, F.; et al. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J. Cell Biol. 2010, 189, 211–221.
- Vives-Bauza, C.; Zhou, C.; Huang, Y.; Cui, M.; de Vries, R.L.; Kim, J.; May, J.; Tocilescu, M.A.; Liu, W.; Ko, H.S.; et al. PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc. Natl. Acad. Sci. USA 2010, 107, 378–383.
- Shiba-Fukushima, K.; Imai, Y.; Yoshida, S.; Ishihama, Y.; Kanao, T.; Sato, S.; Hattori, N. PINK1-mediated phosphorylation of the Parkin ubiquitin-like domain primes mitochondrial translocation of Parkin and regulates mitophagy. Sci. Rep. 2012, 2, 1002.
- Narendra, D.; Tanaka, A.; Suen, D.F.; Youle, R.J. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 2008, 183, 795–803.
- Cho, D.H.; Kim, J.K.; Jo, E.K. Mitophagy and Innate Immunity in Infection. Mol. Cells 2020, 43, 10–22.
- Ham, S.J.; Lee, S.Y.; Song, S.; Chung, J.R.; Choi, S.; Chung, J. Interaction between RING1 (R1) and the Ubiquitin-like (UBL) Domains Is Critical for the Regulation of Parkin Activity. J. Biol. Chem. 2016, 291, 1803–1816.
- Picca, A.; Lezza, A.M.S.; Leeuwenburgh, C.; Pesce, V.; Calvani, R.; Landi, F.; Bernabei, R.; Marzetti, E. Fueling Inflamm-Aging through Mitochondrial Dysfunction: Mechanisms and Molecular Targets. Int. J. Mol. Sci. 2017, 18, 933.
- Picca, A.; Calvani, R.; Coelho-Junior, H.J.; Marzetti, E. Cell Death and Inflammation: The Role of Mitochondria in Health and Disease. Cells 2021, 10, 537.
- Missiroli, S.; Genovese, I.; Perrone, M.; Vezzani, B.; Vitto, V.A.M.; Giorgi, C. The Role of Mitochondria in Inflammation: From Cancer to Neurodegenerative Disorders. J. Clin. Med. 2020, 9, 740.
- Shoshan-Barmatz, V.; Shteinfer-Kuzmine, A.; Verma, A. VDAC1 at the Intersection of Cell Metabolism, Apoptosis, and Diseases. Biomolecules 2020, 10, 1485.
- Thinnes, F.P. Opening up of plasmalemma type-1 VDAC to form apoptotic “find me signal” pathways is essential in early apoptosis—Evidence from the pathogenesis of cystic fibrosis resulting from failure of apoptotic cell clearance followed by sterile inflammation. Mol. Genet. Metab. 2014, 111, 439–444.
- Karachitos, A.; Jordan, J.; Kmita, H. VDAC-Targeted Drugs Affecting Cytoprotection and Mitochondrial Physiology in Cerebrovascular and Cardiovascular Diseases. Curr. Med. Chem. 2017, 24, 4419–4434.
- Shoshan-Barmatz, V.; Mizrachi, D. VDAC1: From structure to cancer therapy. Front. Oncol. 2012, 2, 164.
- Shoshan-Barmatz, V.; Nahon-Crystal, E.; Shteinfer-Kuzmine, A.; Gupta, R. VDAC1, mitochondrial dysfunction, and Alzheimer’s disease. Pharmacol. Res. 2018, 131, 87–101.
- Liu, Y.; Ma, X.; Fujioka, H.; Liu, J.; Chen, S.; Zhu, X. DJ-1 regulates the integrity and function of ER-mitochondria association through interaction with IP3R3-Grp75-VDAC1. Proc. Natl. Acad. Sci. USA 2019, 116, 25322–25328.
- Sasaki, K.; Donthamsetty, R.; Heldak, M.; Cho, Y.E.; Scott, B.T.; Makino, A. VDAC: Old protein with new roles in diabetes. Am. J. Physiol. Cell Physiol. 2012, 303, C1055–C1060.
- Li, X.; Pan, J.; Li, H.; Li, G.; Liu, B.; Tang, X.; Liu, X.; He, Z.; Peng, Z.; Zhang, H.; et al. DsbA-L interacts with VDAC1 in mitochondrion-mediated tubular cell apoptosis and contributes to the progression of acute kidney disease. EBioMedicine 2022, 76, 103859.
- Nowak, G.; Megyesi, J.; Craigen, W.J. Deletion of VDAC1 Hinders Recovery of Mitochondrial and Renal Functions After Acute Kidney Injury. Biomolecules 2020, 10, 585.
- Shoshan-Barmatz, V.; Anand, U.; Nahon-Crystal, E.; Di Carlo, M.; Shteinfer-Kuzmine, A. Adverse Effects of Metformin From Diabetes to COVID-19, Cancer, Neurodegenerative Diseases, and Aging: Is VDAC1 a Common Target? Front. Physiol. 2021, 12, 730048.


