Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Irshad Ahmad | -- | 4410 | 2022-10-20 06:07:34 | | | |
| 2 | Sirius Huang | Meta information modification | 4410 | 2022-10-20 07:35:33 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Ahmad, I. CRISPR/Cas9 as a Promising Tool to Cure Blindness. Encyclopedia. Available online: https://encyclopedia.pub/entry/30310 (accessed on 25 July 2026).
Ahmad I. CRISPR/Cas9 as a Promising Tool to Cure Blindness. Encyclopedia. Available at: https://encyclopedia.pub/entry/30310. Accessed July 25, 2026.
Ahmad, Irshad. "CRISPR/Cas9 as a Promising Tool to Cure Blindness" Encyclopedia, https://encyclopedia.pub/entry/30310 (accessed July 25, 2026).
Ahmad, I. (2022, October 20). CRISPR/Cas9 as a Promising Tool to Cure Blindness. In Encyclopedia. https://encyclopedia.pub/entry/30310
Ahmad, Irshad. "CRISPR/Cas9 as a Promising Tool to Cure Blindness." Encyclopedia. Web. 20 October, 2022.
Copy Citation
CRISPR/Cas9 has been explored as an efficient therapeutic tool for the treatment of genetic diseases. It has been widely used in ophthalmology research by using mouse models to correct pathogenic mutations in the eye stem cells. CRISPR/Cas9 has been used to correct a large number of mutations related to inherited retinal disorders. In vivo therapeutic advantages for retinal diseases have been successfully achieved in some rodents. Advances in the CRISPR-based gene-editing domain, such as modified Cas variants and delivery approaches have optimized its application to treat blindness.
CRISPR
retinal degeneration
eye diseases
blindness
eye therapeutics
viral vectors
non-viral vectors
1. CRISPR-Cas System: Development, Components, and Mechanism
In 1987, scientists have observed a rare structure comprising 29 vastly conserved nucleotides located in the 3′-end adjoining region of the iap gene in E. coli. Afterward analogous repeats have been found in many strains of bacteria and archaea by random sequencing of their complete genomes [1]. These sequences of a clustered repeat were termed short regular spaced repeats (SRSRs) that are frequently spaced via unique intervening sequences of constant length and have been classified as an exclusive gene family existing in the immune systems of prokaryotes providing them an acquired resistance against viruses [2]. The existence of these inward short, inverted repeats in the repetitive units shows the same physiognomies of known sites intended for specific DNA-binding proteins. Many queries have been raised from these findings, such as whether their existence may be due to some of the earliest sequences or have been diverged through the process of evolution.
At the beginning of the 20th century, this innovative repetitive family ‘CRISPR’ was discovered via in silico analysis by revealing many genes that existed in line with the cluster repeats that were denoted as CRISPR-linked genes or Cas genes. Later on, these gene families of CRISPR were recognized as stably maintained secondary structures of RNA demonstrating efficient preservation through the process of evolution [3]. These structures are considered to perform significant functions in the prokaryotic gene repair and regulation to instigate their defense against different viruses. Subsequently, tremendous applications of these structures have been studied to edit animal and human genes using stem cells in order to generate restrictive mutations to find out the inside working environment of the cell. Certainly, the applications of the CRISPR-Cas9 system seem to be beyond our imagination.
The CRISPR-Cas9 system was first investigated in prokaryotes which are using it as an adaptive immune system to counter phage viruses. The system comprises two important constituents, e.g., an endonuclease (Cas9) and a sole guide RNA (sgRNA) used to detect a specific sequence in the genome. The Cas9 act as an endonuclease by cleaving a specific sequence in the genome containing two catalytical positions (RuvC and HNH), nuclease domains that produce DSBs by cleaving the reverse strands of DNA [4]. SgRNA contains two components to enhance its action that comprises CRISPR RNA (crRNA) and an intergenic trans-activating crRNA (tracrRNA) used as a guided tool required for the activity of Cas9 activity. The crRNA contains a sequence ~20 bp that binds to its target sequence following the complementary base pairing rules of Watson–Crick model. The crRNA recognizes the target based on a DNA sequence that is short and conserved called protospacer-adjacent motif (PAM) which is positioned adjacent to the target DNA. SgRNA can be simply designed and cast off rapidly. It unites the Cas9 as a ribonucleoprotein (RNP) complex in the CRISPR structure and SgRNA directs the Cas9 to the targeted position in the anticipated gene and at that point, Cas9 exactly cut the DNA at 2–3 nucleotides earlier than the PAM sequence to generate DSBs. Generally, HDR or NHEJ are the two key mechanisms in DNA repair therefore DSBs can be repaired by any of these mechanisms. Similarly, the sgRNA may be restructured by modest variations in the DNA sequence to retarget novel DNA sequences. Furthermore, the CRISPR-Cas9 system can be used to change numerous genes through a solitary nuclease employing diverse sgRNAs simultaneously (Figure 1).
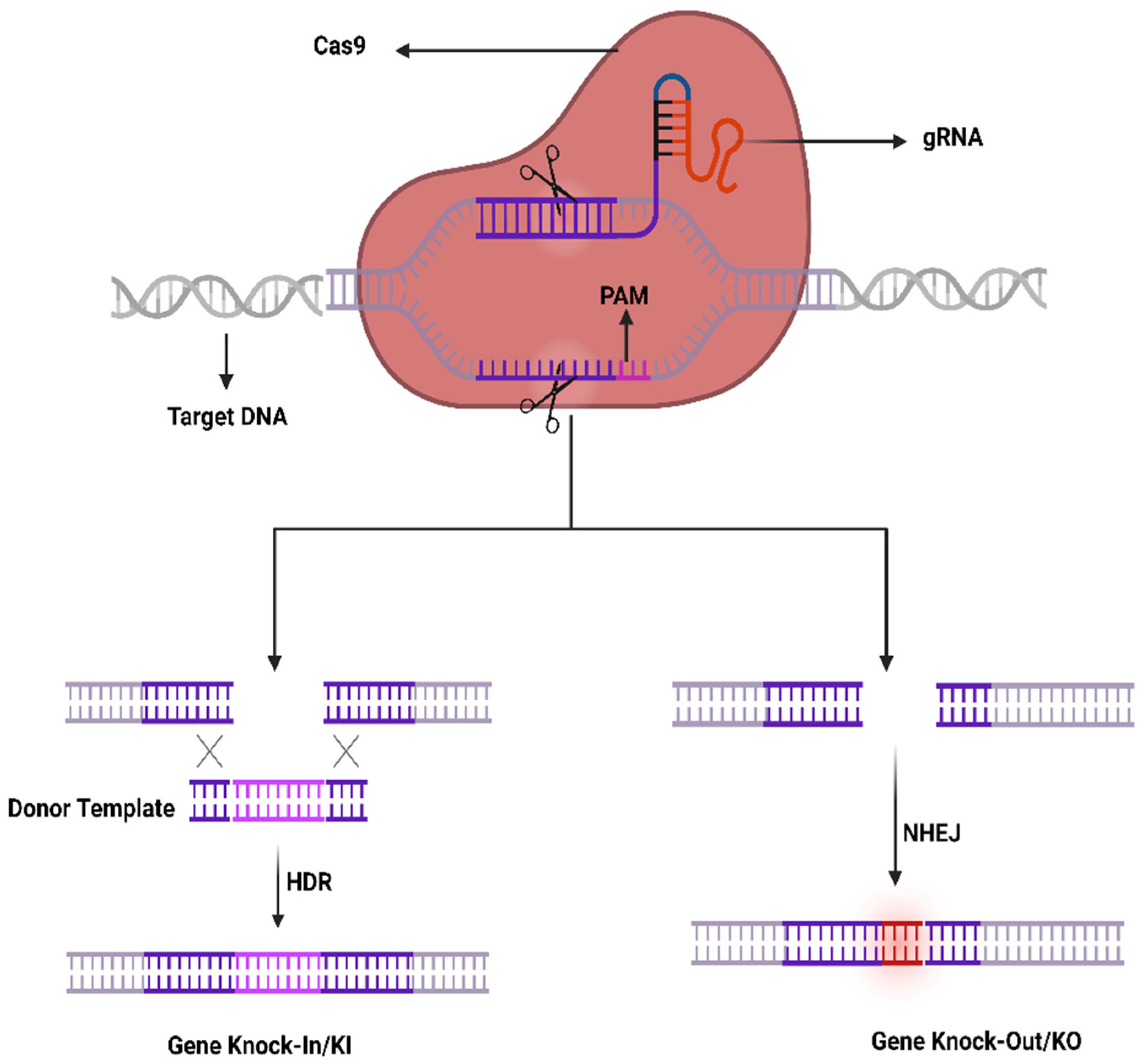
Figure 1. CRISPR/Cas9 working mechanism.
An alternative form of Cas 9 is Cas9 nickase (nCas9) which varies in a single point mutation at position D10A or H840A as of Cas9 and cuts single-stranded DNA sequences targeted by sgRNA. Likewise, the double mutation in H840A and D10A of the HNH and RuvC domains of Cas9 may disable the endonuclease action of Cas9 and can reinforce dead Cas9 (dCas9) [5] which are unable to cut the DNA positions in order to generate DSBs; Therefore, it may be attached to transcriptional activators to make a CRISPR activation (CRISPRa) system capable of altering the gene expression by rewriting the epigenetic marks on the target gene; moreover, It may be attached with transcriptional inhibitors to develop a CRISPR inhibition (CRISPRi) system used to overwhelm the process of transcription [6].
2. CRISPR-Cas System Components Delivery to Ocular Tissues
Different types of injection routes have been used to deliver to ocular cells such as topical administration, intracameral injection, and subconjunctival, that are commonly used for gene transfer, and that can be used for the transfer of CRISPR/Cas9 components. The plasmids DNA gene has been delivered for transgene expression through topical administration. The intracameral injection is usually used to transfer the genes to the interior part of ocular cells such as the Conroy endothelial, and ciliary body. The intravitreal rout injections are used to transfer the genes to the inner retina cells, especially into the ganglion cells. The subretinal injections have been used successfully to transfer transgenes to the outer parts of the retina. Multi variants types of vectors have been designed and used to transfer the Cas9 and RNA components to targeted ocular cells. The Cas9 as of Streptococcus pyogenes (SpCas9) is considered as the most studied ortholog however, due to its greater size (4.2 kb), confining its capacity for in vivo use to be packed with gRNAs into a single adeno-associated virus (AAV) vector. To overcome this constraint, packaging into lentiviral vectors can be employed for twin AAV vectors to express the whole SpCas9 distinctly as of gRNAs, or to use the split-Cas9 by separating the SpCas9 expression on its messy element V713–D718 and remaking the entire protein via trans-splicing of split-intein protein [7]. Furthermore, SaCas9 of Staphylococcus aureus and CjCas9 of Campylobacter jejuni contain minor Cas9 orthologs that can be combined and accompanied by gRNAs simultaneously in the AAV vector [8]. The first time the CRISPR technology was used in the human eye was in the year 2019. The clinical test has assessed the subretinal AAV-facilitated transport of SaCas9 combined with a couple of gRNAs to mark a profound intronic mutation in the CEP290 gene in order to treat type 10 Leber congenital amaurosis (NCT03872479) [9]. Remarkably, even with the theoretical gain to use solitary viral vector for clinical translation, different researchers have observed that genome editing efficacy of the smaller Cas9 alternatives is mediocre with regards to the dual-vector delivery of SpCas9 and gRNAs [8].
Even though viral vectors can be efficiently used as genome editing tools in the retinal cells, the constant viral expression of Cas9 endonuclease may cause off-target effects. In contrast to conservative gene expression or biofactory approaches, the CRISPR systems no longer entail enduring transgene expression, wherever the continued existence of Cas9 may activate indifferent mutations. A different approach intended for clinical use is the straight delivery of recombinant Cas9 proteins along with gRNAs as ribonucleoprotein complexes (RNPs) in the eye that can instantly influence the cleavage of DNA to be quickly degraded in cells, thereby minimizing the off-target effects as well as cytotoxicity [10]. The straight use of RNPs in human cells proved to be an effective gene cleavage method compared to the transfection of plasmid by means of ~79% on-target mutation with less off-target effects and if carried out in the subretinal region for targeting VEGF in the mouse eyes, it resulted in ~40% decline in a CNV laser-induced model. Regardless of the RNP advantages, the Cas9 protein delivery into the nucleus is still a difficult task, mostly attributable to endosomal entrapment in the cytoplasmic matrix. In this regard, the cell-penetrating peptides (CPPs) can enhance the transport efficacy by ~80% [11]. In summary, due to size limitation capacity, mostly the adenosine-associated viral vector (AAV) has been widely used to transfer Cas9 and gRNA cargo into ocular cells. The AVV has a better balance of efficiency and stability. The non-viral vectors can be used in some circumstances, such as off-target effects and immunogenic reactions. With the rapid development of ocular gene delivery vectors, the advanced tailer-designed vectors will enable more efficient and effective CRISPR/Cas9 delivery.
2.1. CRISPR to Treat AMD
AMD is a multi-genetic ailment and the key source of blindness worldwide. A neovascular form of AMD is the wet AMD caused via irregular growth of choroidal vessels in the macula area of the retina ensuing damage to central vision. The macula is controlling the color vision and bright light actions due to their abundant cone photoreceptors [12]. The excess proliferation of vascular endothelial growth factor (VEGF) causes neovascularization in AMD, therefore anti-VEGF mediators turn out to be an excellent therapy [13]. Recently such types of mediators (aflibercept, bevacizumab, and ranibizumab) can be given as an intravitreal injection to treat wet AMD patients [14]. AMD patients can be treated with the AAV-CRISPR tool designed based on CjCas9 (Campylobacter jejuni) [15][16][17] as well as type-V CRISPR-Cas systems, LbCpcf1 nucleases. The CjCas9 gene with its matching sgRNA sequence and marker gene has been packaged into an AAV vector. CjCas9 delivered through AAV can specifically cut a restricted number of sites in the human or mouse genome that can cause desired mutations in the RPE cells. In this regard, the Vegfa or Hif1a gene in RPE cells can be targeted by CjCas9 to reduce the size of laser-induced choroidal neovascularization and this approach can be developed into in vivo genome editing therapy for AMD. After six weeks an AAV-CjCas9 injection intravitreally has resulted from an Indel efficiency of 22 ± 3% in Vegfa and 31 ± 2% in Hifla genes, respectively. Additionally, the outcomes of the Indels have been observed at the protein level e.g., substantial decline in VEGF-A protein was detected in RPE cells as compared to the control set. Another research group used lipofectamine 2000 for the delivery of sgRNA/Cas9 expressing plasmid with Cas9 RNPs which showed 82 ± 5% and 57 ± 3% of indel in NIH3T3 and ARPE-19 cells, respectively. In this study, Cas9 RNPs have been observed to be highly active w.r.t plasmid on the second day of transfection. After treatment with Cas9 RNPs, a reduction in the VEGF A mRNA and protein levels of 40 ± 8% and 52 ± 9% has been observed in the adult retinal pigment epithelial cells (ARPE). To evaluate the in vivo efficacy, Cy3-characterized RNPs have been delivered through intravitreal injection. After three days of injection, Cy3 dye was accumulated into RPE cells with detection of 25 ± 3% of indel at the delivery site in RPE cells. Furthermore, to mimic wAMD a CNV mouse model has been developed using laser tracked by an injection of Cas9 RNPs in the subretinal region. Three days later 22 ± 5% indel was detected in RPE cells for the VEGFA gene. The treatment with Cas9 RNPs has tremendously condensed the CNV area by 8 ± 4% along with a reduced level of VEGF A protein (Figure 2) [18].
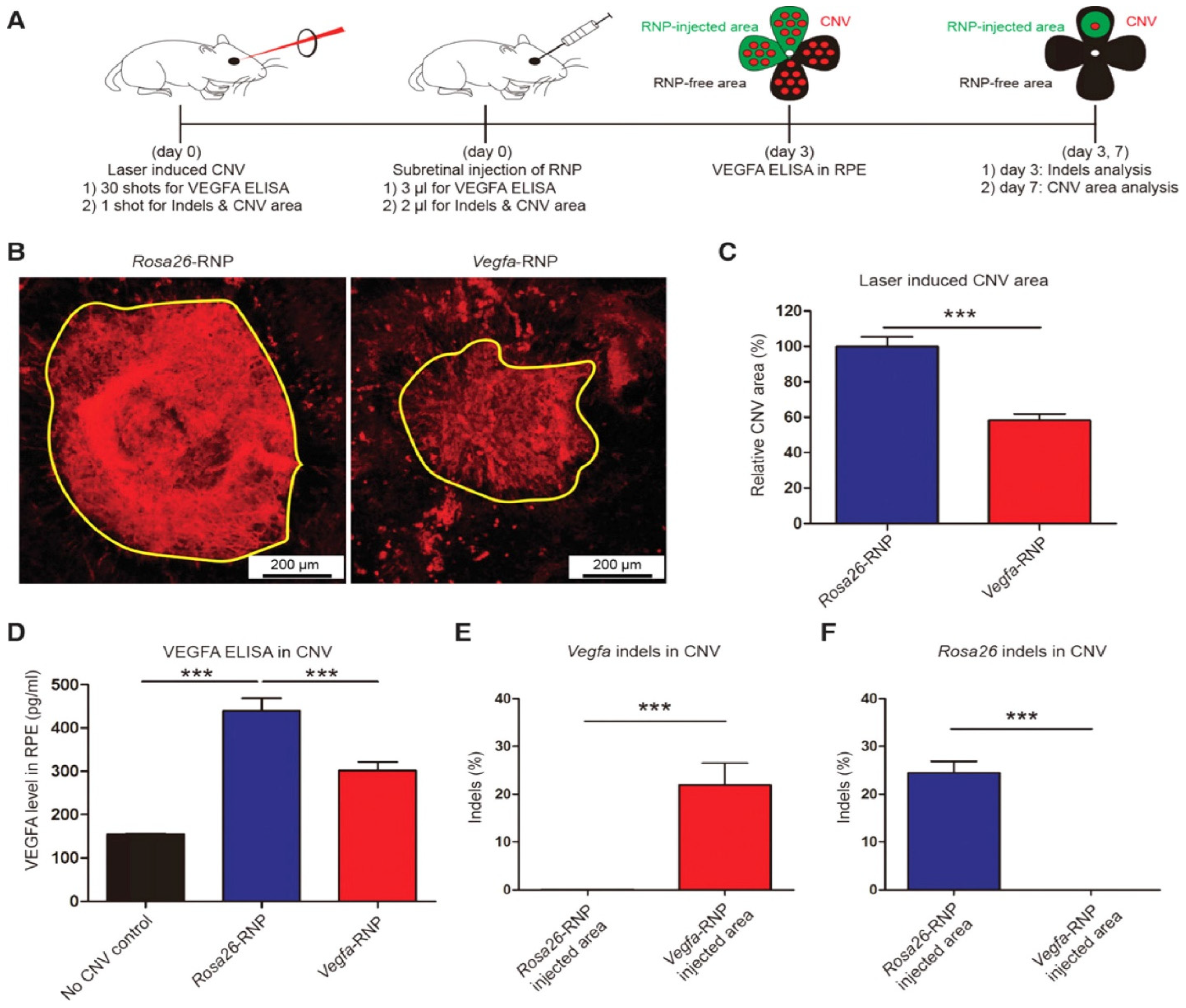
Figure 2. (A) Mice with laser-induced choroidal neovascularization (CNV) injected with Vegfa-specific Cas9 RNP analyzed at 3 days post-injection for Vegfa and 7 days post-injection for INDLES analysis. (B) The laser-induced CNV was stained with isolectin B4. (C) CVN area under Vegfa-RNA. (D) controlled and CRISPR/Cas9 edieted comparioson. (E) INDEL frequencies due to Vegfa-RNA injected. (F) INDEL frequancies at Rosa26 site. Student’s t-test: (***) p < 0.001 [18].
Recently a lentiviral system has been reported to deliver mRNA encoding an extended Cas9 protein (SpCas9) and gRNA instantaneously named mLP-CRISPR. The mLP specifies mRNA-carrying lentiviral particles that are used to prevent the progress of wAMD in a laser-persuaded CNV mouse model. Subretinal injection of mLP-CRISPR displays an enhanced tissue specificity in the retinal pigment epithelium (RPE) cells that are considered to be the main cause of VEGFA in the outer portion of adult eyes. Additionally, mLP-CRISPR did not induce anti-Cas9 IgG in blood or T-cell permeation in the eyes. An injection of mLP-CRISPR has disrupted 44% of Vegfa genes in RPE while decreasing 63% of the laser-induced CNV area in the wAMD mouse model. This has been done by using a gene-editing tool without any off-target effects. This type of mLP technique can be used to carry mRNA encoding numerous Cas9 nucleases, base, prime, and epigenome editors [19].
2.2. CRISPR to Treat the Glaucoma
Glaucoma is an assemblage of eye diseases caused due to advanced and permanent disintegration of the ganglion cells in the retina which axonal projections establish the optic nerve [20]. At present, it is considered the foremost source of irreversible blindness globally [17] that could affect >76 million people by the year 2020. Presently, glaucoma is mainly treated in clinics by reducing the intraocular pressure (IOP) in affected individuals by medicine, laser action, or surgical treatment. However, surgical and laser therapy brings risks that frequently need additional interference or combinational tactics supplemented with more topical therapies during a patient’s lifetime [14]. The patients frequently require consistent treatment through numerous types of eye drops that can be used many times daily. It means that patient treatment becomes a challenge with the passage of time and even numerous of them have experienced continuous visual loss instead of their reduced IOP [21][22]. Consequently, novel therapeutic methods can be developed that can be obtainable by an injection into the eye directly with enduring or perpetual beneficial consequences. A considerable number of adult individuals with glaucoma have an indistinct, varied cause of disease linking numerous genetic, environmental, and individual risk factors [15][23]. Due to these reasons, the commencement of glaucoma in adult individuals can be controlled by gene therapies that are mainly focused on neuroprotection, which encompasses the reduced loss of RGCs by changing their physiological status in order to reduce the severity of the disease. This can be achieved by either enhancing the action of innate survival pathways in RGC or inhibiting the development of programmed cell death.
With the recent developments in specificity and efficacy of CRISPR-mediated genome editing technology, there is a hope to target these mutations. Currently, researchers have targeted the dominant MYOC mutations in a mouse model of myocilin-linked POAG through adenoviruses that are expressing the CRISPR/Cas9 components (Ad5-cas9 and Ad5-crMYOC). In this study, Cas9 has successfully knock-out the mutant MYOC gene, reduced IOP in the eyes of treated mice, and stopped further glaucomatous damage in mouse eyes. Similarly, the same constructs have been used for the treatment of trabecular meshwork tissue in the human eyes cultured ex vivo. A decrease in the myocilin mRNA suggests the possibility of using this technology to overcome the problems of patients with MYOC mutations [24].
Primary open-angle glaucoma (POAG) has been considered the main global source of blindness, comprising an imperative risk factor of elevated intraocular pressure (IOP). The pathological variations in the trabecular meshwork (TM) increase POAG IOP linked with changes caused by an enhanced level of TGFβ2. Recently, CRISPR interference has been used to explicitly deacetylate histones in order to decline TGFβ2 in the TM. The CRISPR interference system has been observed to constrain TGFβ2 expression in human TM cells through accurately designing sgRNA that have targeted the TGFβ2 gene promoter. The sgRNA has targeted the CMV promoter of the Ad5-CMV-TGFβ2 viral vector and it has been observed that lentivirus-mediated KRAB-dCAS9 and sgRNA expression were capable to constrain Ad5-CMV-TGFβ2-induced OHT in the eyes of C57BL/6J mice (male and female). The reduction in OHT was linked with a diminished level of TGFβ2 and extracellular matrix proteins in the mouse eye. These findings suggest that CRISPR interference can be used as a tool for gene inhibition containing the therapeutic potential for the treatment of TGFβ2-induced OHT [25].
Currently, a practical gene therapy tool has been used to reduce IOP via specific disruption of the aqueous humor synthesis in the ciliary body ensuing an intravitreal injection. In this regard, explicit gene editing was done by means of the smaller S. aureus-derived CRISPR-Cas9 that is proficient to be carried in a single recombinant AAV vector which is considered a benchmark by US-FDA and has been approved for visual gene therapy. In order to target a gene vital for a well-maintained functional development relatively to correct a single explicit mutation, a collective approach deprived of excessive personalized tactic is manageable. IOP decrease can be accomplished through knock outing Aquaporin 1 (Aqp1) in the ciliary body. Aquaporins are a group of water-transferring transmembrane proteins that are broadly expressed all over the human body and transgenic mice lacking Aqp1 have shown lesser IOP due to abridged influx to form aqueous humor deprived of contrary consequences [26].
2.3. CRISPR to Treat Retinitis Pigmentosa (RP)
RP is considered an important source of progressive blindness affecting 1 in 4000 individuals [27]. The rod-cone dystrophy is a usual RP that is recognized via shaft vision ultimately leading to the loss of peripheral vision. Its early symptoms contain nyctalopia which leads to night blindness and patients have complications in adjusting to the dark that happened to the injury of rod function in early childhood [4]. Due to the damaged photoreceptors, RPE begins to lose its pigment eventually leas to the buildup of intraretinal melanin deposits, which appeared as a bone spicule conformation. Though, the central vision is still integral till the last stages. Different genetic modes are responsible for its transmission, such as autosomal dominant and recessive or X-linked and are heterogeneously linked with mutations as a minimum of 79 genes [28]. There are primarily two types of RP which are MERTK connected and RPGR X-linked. The apical membrane of RPE comprises photoreceptors that are sensitive to light and mediated by MERTK (Mer tyrosine kinase) receptors that are engaged in the rods and cones phagocytosis. To play an effective role, the continuous recycling of these photoreceptors is very important that is interrupted by mutations in MERTK, which proceed to degradation and ultimately decline of the photoreceptors [29]. It has been observed in meta-analyses that ~3% of MERTK type RP are a result of autosomal recessive transmission which are causing macular atrophy and childhood photoreceptor abnormality [30][31]. Alterations in the RP GTPase regulator (RPGR) that is an X-linked RP (XLRP) has been observed in 1 out of 3500 individuals. RPGR, as well as the δ subunit of rod cGMP phosphodiesterase, controls the proteins and its disorder is causing continuous damage to the central vision and progressing to night blindness [4].
CRISPR/Cas9 technology has been used in some in vitro and in vivo studies to treat RP e.g., applied in a rat model of adRP to remove mutation in the rhodopsin gene (RhoS334). In this experiment the sgRNA/Cas9 plasmid has been used to target exon 1 closely upstream of a PAM exclusive toward the RhoS334 locus was directed intravitreally in S334ter-3 rats. In two different rats, a cleavage efficacy of 33 and 36% was confirmed in transfected retinal cells through genome analysis. Subsequent injection of sgRNA/Cas9 plasmid has confirmed an enhanced visual insight and widespread protection of the retina through immunohistological examination [32]. Moreover, the same strategy was used to edit the RHO gene mutations. In this research, an intended plasmid containing an insert for 2 sgRNAs targeting the RHO gene was used to generate DSB and subsequently NHEJ. As a result of this research, the RHO gene was successfully edited with additional downregulation of the expressing RHO protein. In another study, CRISPR/Cas9 system was used in iPSCs attained from a patient with photoreceptor degeneration to treat XLRP to correct RPGR (a pathogenic point mutation). In this research, 21 different sgRNAs have been screened for editing, and among them, g58 was observed as utmost activity. Thus, the plasmid containing g58/Cas9 was considered to transfect the iPSCs together with RPGR single-stranded oligo deoxy ribonucleotide (ssODN) that plays a supportive role in the HDR pathway. After deep sequencing analysis, the data showed an effective modification of mutation in 13% of the transfected cells [33]. Additionally, the premature stop codon TAG was successfully substituted by the wild-type codon GAG encoding glutamate at position 1024. In contrast, the untransfected iPSCs did not show any variations in the mentioned mutation. It was determined that the 13% correction rate was meaningfully productive that can be further enhanced by reducing error-prone NHEJ through DNA ligase IV inhibition on the DNA cleavage site. Moreover, a CRISPR/Cas-based approach was established to edit RHO gene mutations in the ADRP mouse model using a plasmid comprising an insert designed for two sgRNAs to target the RHO gene (exon 1) containing P23H dominant mutation. Primarily, HeLa cells have been used for gene editing in vitro showing an indel frequency of 70%, 76%, and 82% by means of sgRNA1, sgRNA3, and 2sgRNA, respectively. Further, the Real-time Taqman PCR has been used to observe RHO expression where 35%, 25%, and 20% of reduced expression level has been detected in the cells treated with sgRNA1, sgRNA3, and 2sgRNA, respectively. Later on, the CRISPR/Cas plasmid has been used by electroporation in P23 RHO transgenic mice comprising 2sgRNA together with green fluorescence protein (GFP) to achieve subretinal expression. To evaluate Cas9 expression, the GFP expressing section of the retina has been isolated where the expression was inadequate in the cells articulating GFP together with 84 edited sequences [33].
The scope of treatment is the inadequate beginning embryonic day (E) E16 to P2 due to the rate of retinal deterioration in adRP patients that is heterogeneous. Therefore, an ablation therapy for long duration could be useful to maintain vision in animal models. Thereby, a slower degenerating adRP model having a common adRP mutation (rhodopsin P23H) has been used for clinical application. In adRP patients, the P23H (RHOP23H) is a very common mutation that can be observed in the rod cell-specific gene rhodopsin (RHO) which is an effective mark to study mutation-specific ablation approaches via CRISPR [34][35]. Usually, an excessive level of rhodopsin misfolding (class II mutation) and mistrafficking (class I mutation) as a result of P23H mutation is accumulated in the endoplasmic reticulum (ER) [36][37][38]. The P23H line-3 rats experience photoreceptor degeneration from P15 which shows the level of vision retained with the passage of time equal to decades in patients accepted for treatment at various disease stages. In this research, the subretinal delivery of AAV-CRISPR/Cas9 has been shown as a benign approach that can cure photoreceptors and vision intended for 15 months in Rhodopsin P23H-3 rats (Figure 3) [39].
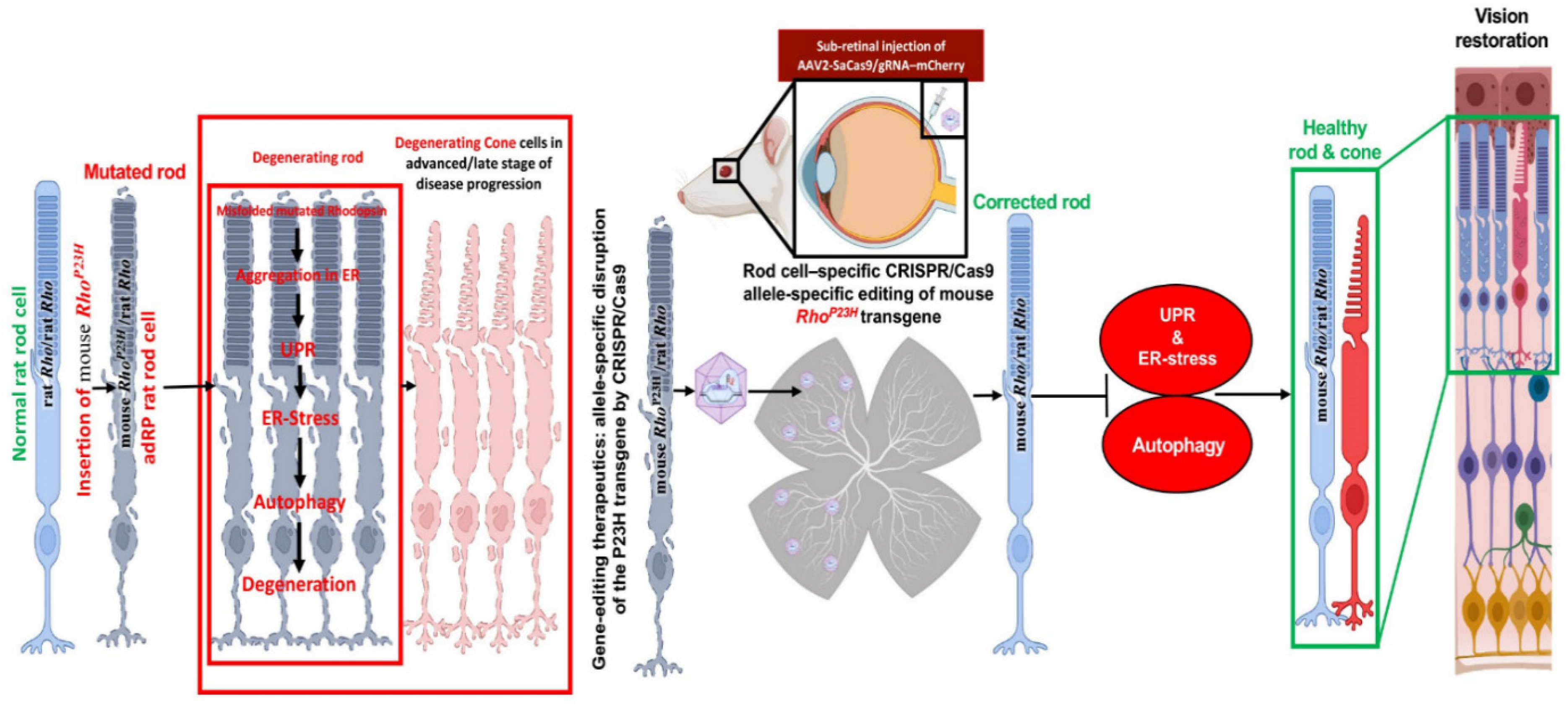
Figure 3. Schematic representation of CRISPR/Cas9 editing in the Rhodopsin P23H to restore and rescue long-term vision in mice [39].
2.4. CRISPR to Treat Leber Congenital Amaurosis (LCA10)
LCA is considered a high overwhelming retinal dystrophy due to its potential of causing congenital blindness in <1 year of age. Until now 14 mutant genes have been known based on linkage examination, homozygosity mapping, and genome scrutiny in LCA patients among which 70% are children with retinal degeneration [40]. LCA patients are generally connected with plain imperfections comprising rambling eye movements termed nystagmus. In children, the symptoms are usually sluggish pupil reactions and deficiency of electroretinographic reactions [5][29]. Genes intricate LCA encode the proteins that are accountable for retinal roles comprising vitamin A cycling (LRAT, RPE65, RDH12), photoreceptor morphogenesis (CRB1, CRX), guanine synthesis (IMPDH1), phototransduction (AIPL1, GUCY2D), outer segment phagocytosis (MERTK), and intra-photoreceptor ciliary transport progressions (CEP290, RPGRIP1, LCA5, TULP1) [40]. Until now, the investigated genes for LCA stand mutations in the RPE (RPE65) gene that is encoding retinoid isomerase [41], although, the maximum going on mutations are connected with the CEP290 (15%), GUCY2D (12%), and CRB1 (10%), respectively. About 20% of north-western Europe patients have an intronic CEP290 mutation (p.Cys998X). In the mice, an AVV-CRISPR system has been studied to investigate the in vivo action of autosomal dominant retinitis pigmentosa (adRP) and LCA10. In this research, the AAV-SpCas9 vector was carried through subretinal injections targeting the rhodopsin (RHO) or CEP290 and Nrl (neural retina leucine zipper transcription factor) gene using a mouse model for adRP. The results are very promising which show an enhanced level of spCas9 protein expression in the retinal cells of mice for a period of nine-and-a-half months. Additionally, the researchers have arrayed diverse AAV serotypes besides changed vector doses and have found a real repair of RP or LCA10 phenotype lacking off-target properties or adverse toxic responses [30][31]. The same approach has been used successfully in human cells to resolve the RHO gene mutation. This study confirms CRISPR/Cas9 as an effective system to target gene/alleles in a well-organized way indicating that it may be cast off for the treatment of RP and further dominant human genetic disorders [42].
2.5. CRISPR to Treat the Usher Syndrome
Usher syndrome is considered a frequent form of syndromic IRD with a prevalence of 1 out of 20,000 individuals with its exclusive characteristics comprising RP and hearing damage [16]. Based on the development and intensity of the hearing injury along with the RP outset, this heterogenous syndrome is segregated into three subtypes which are usher syndrome type 1 (USH1) utmost critical, USH2 frequently observed with modest to severe symptoms, and USH3 considered as a moderate phenotype that varies case by case regarding disease outset and its progression [17]. USH1 is considered to be the main source of deaf-blindness that is inherited in an autosomal recessive manner in humans causing vestibular ailment, deep congenital heart loss, and RP. USH1 is triggered as a result of mutations in myosin VIIA encoding an organelle transport protein in the RPE [13]. CRISPR/Cas9 gene editing was proposed as a gene-editing tool to reinstate the c.2299delG mutation in the USH2A gene. The gene-editing experiment was carried out in human dermal fibroblasts (HDFs) cells taken from a USH2 patient having c.2299delG mutation. By means of nucleofection, a Cas9 RNPs containing 15 µg Cas9 and 20 µg sgRNA has been transfected into the normal individual HDFs producing 18% indel rates. Afterward, delivery of RNPs combined with ssODN-2299 has produced 5% HDR efficacy. Correspondingly, patient HDFs with c.2299delG mutation have been transfected with ssODN having a WT sequence with a deleted PAM sequence resulting in 6% indel efficacy with 2.5% deletion of HDR [18].
Recently a pig model of USH imitates the mix of deafness, vestibular dysfunction, and vision loss found in USH individuals. Visual impairment has been observed in USH1C pigs through ophthalmologic checkups, studying their behavioral examinations go together with morphologic changes in the photoreceptor cells. Additionally, the photoreceptor and primary cilia of fibroblasts have been constantly extended in USH1C pigs which could help to conduct gene-editing experiments as potential therapeutics. Investigative gene therapeutic tools can verify the improvement of visual function in USH1C pigs (Figure 4) [43].
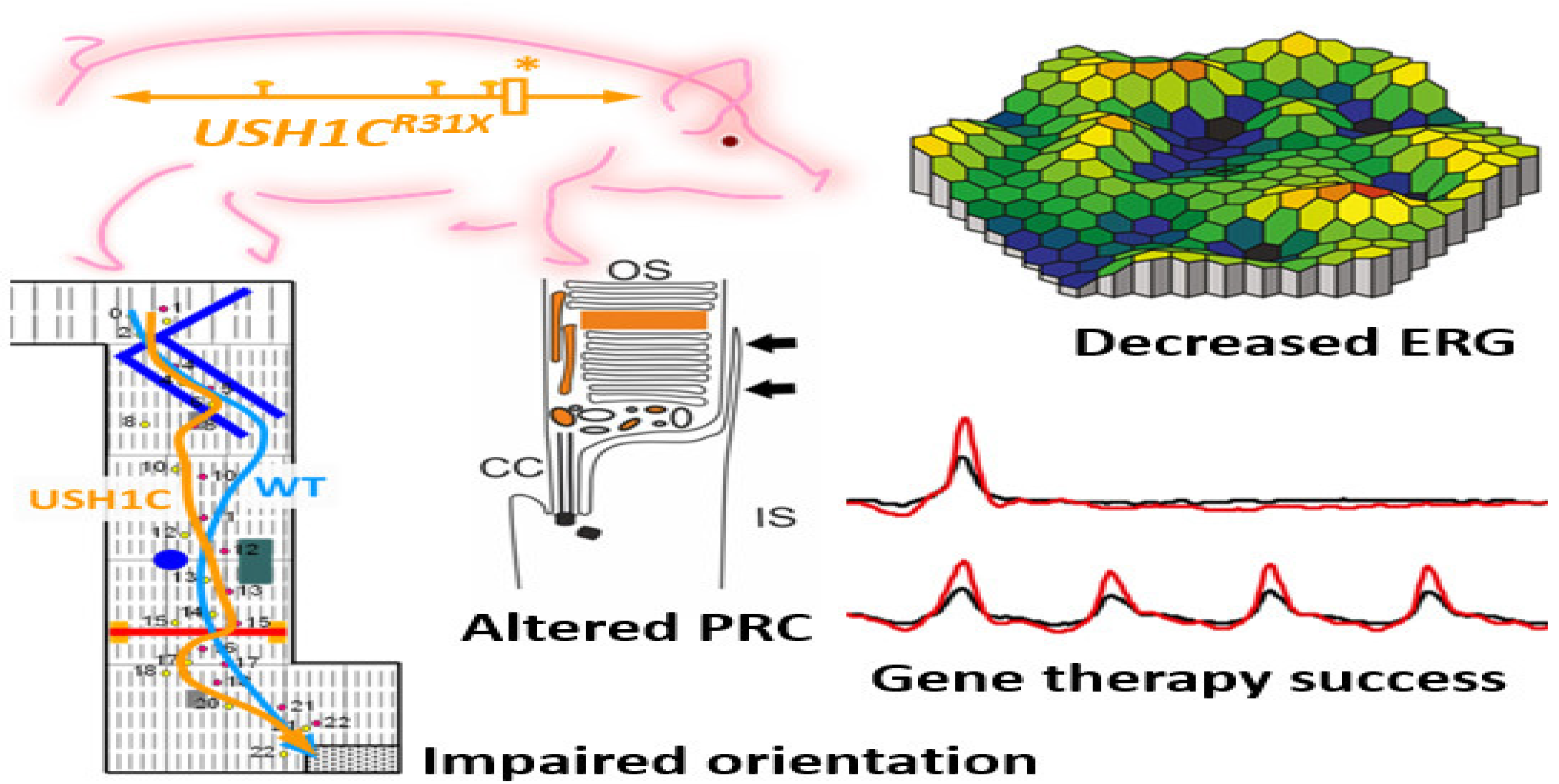
Figure 4. Schematic representation of disruption of photoreceptors architecture to rectify usher syndrome through CRISPR/Cas9 to restore the vision in pig. * Denotes the R31X disruptive mutation at USH gene in the pig model. ERG recordings in USH1C mutant pigs vs. WT demonstrate a significant reduction in the scotopic standard flash responses, dark is dim light and red with bright light stimulus. Matrix plot denotes the impaired orientation of mutant and wild type pigs [43].
References
- Jansen, R.; Embden, J.D.v.; Gaastra, W.; Schouls, L.M. Identification of genes that are associated with DNA repeats in prokaryotes. Mol. Microbiol. 2002, 43, 1565–1575.
- Mojica, F.J.; Díez-Villaseñor, C.; García-Martínez, J.; Almendros, C. Short motif sequences determine the targets of the prokaryotic CRISPR defence system. Microbiology 2009, 155, 733–740.
- Kunin, V.; Sorek, R.; Hugenholtz, P. Evolutionary conservation of sequence and secondary structures in CRISPR repeats. Genome Biol. 2007, 8, R61.
- Scholl, H.P.; Strauss, R.W.; Singh, M.S.; Dalkara, D.; Roska, B.; Picaud, S.; Sahel, J.-A. Emerging therapies for inherited retinal degeneration. Sci. Transl. Med. 2016, 8, rv366–rv368.
- Buecher, B.; Gauthier-Villars, M.; Desjardins, L.; Rouic, L.-L.; Levy, C.; De Pauw, A.; Bombled, J.; Tirapo, C.; Houdayer, C.; Bressac-de Paillerets, B. Contribution of CDKN2A/P16 INK4A, P14 ARF, CDK4 and BRCA1/2 germline mutations in individuals with suspected genetic predisposition to uveal melanoma. Fam. Cancer 2010, 9, 663–667.
- Nakade, S.; Yamamoto, T.; Sakuma, T. Cas9, Cpf1 and C2c1/2/3—What’s next? Bioengineered 2017, 8, 265–273.
- Chew, W.L.; Tabebordbar, M.; Cheng, J.K.; Mali, P.; Wu, E.Y.; Ng, A.H.; Zhu, K.; Wagers, A.J.; Church, G.M. A multifunctional AAV–CRISPR–Cas9 and its host response. Nat. Methods 2016, 13, 868–874.
- Chung, S.H.; Mollhoff, I.N.; Nguyen, U.; Nguyen, A.; Stucka, N.; Tieu, E.; Manna, S.; Meleppat, R.K.; Zhang, P.; Nguyen, E.L. Factors impacting efficacy of AAV-mediated CRISPR-based genome editing for treatment of choroidal neovascularization. Mol. Ther.-Methods Clin. Dev. 2020, 17, 409–417.
- Maeder, M.L.; Stefanidakis, M.; Wilson, C.J.; Baral, R.; Barrera, L.A.; Bounoutas, G.S.; Bumcrot, D.; Chao, H.; Ciulla, D.M.; DaSilva, J.A. Development of a gene-editing approach to restore vision loss in Leber congenital amaurosis type 10. Nat. Med. 2019, 25, 229–233.
- Kim, J.; Hwang, J.-M.; Ko, H.; Seong, M.-W.; Park, B.-J.; Park, S. Mitochondrial DNA content is decreased in autosomal dominant optic atrophy. Neurology 2005, 64, 966–972.
- Zuris, J.A.; Thompson, D.B.; Shu, Y.; Guilinger, J.P.; Bessen, J.L.; Hu, J.H.; Maeder, M.L.; Joung, J.K.; Chen, Z.-Y.; Liu, D.R. Efficient delivery of genome-editing proteins in vitro and in vivo. Nat. Biotechnol. 2015, 33, 73.
- Petit, L.; Khanna, H.; Punzo, C. Advances in gene therapy for diseases of the eye. Hum. Gene Ther. 2016, 27, 563–579.
- Yue, L.; Weiland, J.D.; Roska, B.; Humayun, M.S. Retinal stimulation strategies to restore vision: Fundamentals and systems. Prog. Retin. Eye Res. 2016, 53, 21–47.
- Ayton, L.N.; Barnes, N.; Dagnelie, G.; Fujikado, T.; Goetz, G.; Hornig, R.; Jones, B.W.; Muqit, M.K.; Rathbun, D.L.; Stingl, K.; et al. An update on retinal prostheses. Clin. Neurophysiol. 2020, 131, 1383–1398.
- Liu, K.; Lai, T.Y.; Chiang, S.W.; Chan, V.C.; Young, A.L.; Tam, P.O.; Pang, C.P.; Chen, L.J. Gender specific association of a complement component 3 polymorphism with polypoidal choroidal vasculopathy. Sci. Rep. 2014, 4, 7018.
- Kuo, J.Z.; Wong, T.Y.; Ong, F.S. Genetic risk, ethnic variations and pharmacogenetic biomarkers in AMD and polypoidal choroidal vasculopathy. Expert Rev. Ophthalmol. 2013, 8, 127–140.
- Imamura, Y.; Engelbert, M.; Iida, T.; Freund, K.B.; Yannuzzi, L.A. Polypoidal choroidal vasculopathy: A review. Surv. Ophthalmol. 2010, 55, 501–515.
- Kim, K.; Park, S.W.; Kim, J.H.; Lee, S.H.; Kim, D.; Koo, T.; Kim, K.-e.; Kim, J.H.; Kim, J.-S. Genome surgery using Cas9 ribonucleoproteins for the treatment of age-related macular degeneration. Genome Res. 2017, 27, 419–426.
- Ling, S.; Yang, S.; Hu, X.; Yin, D.; Dai, Y.; Qian, X.; Wang, D.; Pan, X.; Hong, J.; Sun, X. Lentiviral delivery of co-packaged Cas9 mRNA and a Vegfa-targeting guide RNA prevents wet age-related macular degeneration in mice. Nat. Biomed. Eng. 2021, 5, 144–156.
- Ma, M.; Li, Z.; Wang, D.W.; Wei, X. Next-generation sequencing identifies novel mutations in the FBN1 gene for two Chinese families with Marfan syndrome. Mol. Med. Rep. 2016, 14, 151–158.
- Brown, B. FDA Approves Upgraded System for the Blind. 2021. Available online: https://healthtechinsider.com/2021/03/08/fda-approves-upgraded-system-for-the-blind/ (accessed on 8 March 2021).
- Nanegrungsunk, O.; Au, A.; Sarraf, D.; Sadda, S.R. New frontiers of retinal therapeutic intervention: A critical analysis of novel approaches. Ann. Med. 2022, 54, 1067–1080.
- Laude, A.; Cackett, P.D.; Vithana, E.N.; Yeo, I.Y.; Wong, D.; Koh, A.H.; Wong, T.Y.; Aung, T. Polypoidal choroidal vasculopathy and neovascular age-related macular degeneration: Same or different disease? Prog. Retin. Eye Res. 2010, 29, 19–29.
- Jain, A.; Zode, G.; Kasetti, R.B.; Ran, F.A.; Yan, W.; Sharma, T.P.; Bugge, K.; Searby, C.C.; Fingert, J.H.; Zhang, F. CRISPR-Cas9–based treatment of myocilin-associated glaucoma. Proc. Natl. Acad. Sci. USA 2017, 114, 11199–11204.
- Rayana, N.P.; Sugali, C.K.; Dai, J.; Peng, M.; Liu, S.; Zhang, Y.; Wan, J.; Mao, W. Using CRISPR Interference as a Therapeutic Approach to Treat TGFβ2-Induced Ocular Hypertension and Glaucoma. Investig. Ophthalmol. Vis. Sci. 2021, 62, 7.
- Wu, J.; Bell, O.H.; Copland, D.A.; Young, A.; Pooley, J.R.; Maswood, R.; Evans, R.S.; Khaw, P.T.; Ali, R.R.; Dick, A.D. Gene therapy for glaucoma by ciliary body aquaporin 1 disruption using CRISPR-Cas9. Mol. Ther. 2020, 28, 820–829.
- Beauchamp, M.S.; Oswalt, D.; Sun, P.; Foster, B.L.; Magnotti, J.F.; Niketeghad, S.; Pouratian, N.; Bosking, W.H.; Yoshor, D. Dynamic Stimulation of Visual Cortex Produces Form Vision in Sighted and Blind Humans. Cell 2020, 181, 774–783.
- Coupland, S.E.; Lake, S.L.; Zeschnigk, M.; Damato, B. Molecular pathology of uveal melanoma. Eye 2013, 27, 230–242.
- Werdich, X.Q.; Jakobiec, F.A.; Singh, A.D.; Kim, I.K. A review of advanced genetic testing for clinical prognostication in uveal melanoma. Semin. Ophthalmol. 2013, 28, 361–371.
- Cohn, A.C.; Toomes, C.; Hewitt, A.W.; Kearns, L.S.; Inglehearn, C.F.; Craig, J.E.; Mackey, D.A. The natural history of OPA1-related autosomal dominant optic atrophy. Br. J. Ophthalmol. 2008, 92, 1333–1336.
- Toomes, C.; Marchbank, N.J.; Mackey, D.A.; Craig, J.E.; Newbury-Ecob, R.A.; Bennett, C.P.; Vize, C.J.; Desai, S.P.; Black, G.C.; Patel, N. Spectrum, frequency and penetrance of OPA1 mutations in dominant optic atrophy. Hum. Mol. Genet. 2001, 10, 1369–1378.
- Bakondi, B.; Lv, W.; Lu, B.; Jones, M.K.; Tsai, Y.; Kim, K.J.; Levy, R.; Akhtar, A.A.; Breunig, J.J.; Svendsen, C.N. In vivo CRISPR/Cas9 gene editing corrects retinal dystrophy in the S334ter-3 rat model of autosomal dominant retinitis pigmentosa. Mol. Ther. 2016, 24, 556–563.
- Xu, C.L.; Park, K.S.; Tsang, S.H. CRISPR/Cas9 genome surgery for retinal diseases. Drug Discov. Today Technol. 2018, 28, 23–32.
- Ayala-Haedo, J.A.; Gallins, P.J.; Whitehead, P.L.; Schwartz, S.G.; Kovach, J.L.; Postel, E.A.; Agarwal, A.; Wang, G.; Haines, J.L.; Pericak-Vance, M.A. Analysis of Single Nucleotide Polymorphisms in the NOS2A Gene and Interaction with Smoking in Age-Related Macular Degeneration. Ann. Hum. Genet. 2010, 74, 195–201.
- Qi, J.H.; Bell, B.; Singh, R.; Batoki, J.; Wolk, A.; Cutler, A.; Prayson, N.; Ali, M.; Stoehr, H.; Anand-Apte, B. Sorsby Fundus Dystrophy Mutation in Tissue Inhibitor of Metalloproteinase 3 (TIMP3) promotes Choroidal Neovascularization via a Fibroblast Growth Factor-dependent Mechanism. Sci. Rep. 2019, 9, 17429.
- Liutkeviciene, R.; Vilkeviciute, A.; Gedvilaite, G.; Kaikaryte, K.; Kriauciuniene, L. Haplotypes of HTRA1 rs1120638, TIMP3 rs9621532, VEGFA rs833068, CFI rs10033900, ERCC6 rs3793784, and KCTD10 rs56209061 Gene Polymorphisms in Age-Related Macular Degeneration. Dis. Markers 2019, 2019, 9602949.
- Vishal, M.; Sharma, A.; Kaurani, L.; Alfano, G.; Mookherjee, S.; Narta, K.; Agrawal, J.; Bhattacharya, I.; Roychoudhury, S.; Ray, J. Genetic association and stress mediated down-regulation in trabecular meshwork implicates MPP7 as a novel candidate gene in primary open angle glaucoma. BMC Med. Genom. 2016, 9, 15.
- Thorleifsson, G.; Walters, G.B.; Hewitt, A.W.; Masson, G.; Helgason, A.; DeWan, A.; Sigurdsson, A.; Jonasdottir, A.; Gudjonsson, S.A.; Magnusson, K.P. Common variants near CAV1 and CAV2 are associated with primary open-angle glaucoma. Nat. Genet. 2010, 42, 906–909.
- Shahin, S.; Xu, H.; Lu, B.; Mercado, A.; Jones, M.K.; Bakondi, B.; Wang, S. AAV-CRISPR/Cas9 Gene Editing Preserves Long-Term Vision in the P23H Rat Model of Autosomal Dominant Retinitis Pigmentosa. Pharmaceutics 2022, 14, 824.
- Metz, C.; Lohmann, D.; Zeschnigk, M.; Bornfeld, N. Uveal melanoma: Current insights into clinical relevance of genetic testing. Klin. Mon. Fur Augenheilkd. 2013, 230, 686–691.
- Al-Maskari, A.; O’grady, A.; Pal, B.; McKibbin, M. Phenotypic progression in X-linked retinitis pigmentosa secondary to a novel mutation in the RPGR gene. Eye 2009, 23, 519–521.
- Gumerson, J.D.; Alsufyani, A.; Yu, W.; Lei, J.; Sun, X.; Dong, L.; Wu, Z.; Li, T. Restoration of RPGR expression in vivo using CRISPR/Cas9 gene editing. Gene Ther. 2022, 29, 81–93.
- Grotz, S.; Schäfer, J.; Wunderlich, K.A.; Ellederova, Z.; Auch, H.; Bähr, A.; Runa-Vochozkova, P.; Fadl, J.; Arnold, V.; Ardan, T. Early disruption of photoreceptor cell architecture and loss of vision in a humanized pig model of usher syndromes. EMBO Mol. Med. 2022, 14, e14817.
More
Information
Subjects:
Genetics & Heredity
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.1K
Revisions:
2 times
(View History)
Update Date:
20 Oct 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No