 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | André T. S. Vicente | -- | 3180 | 2022-10-12 17:22:44 | | | |
| 2 | Camila Xu | Meta information modification | 3180 | 2022-10-14 04:32:08 | | | | |
| 3 | Camila Xu | Meta information modification | 3180 | 2022-10-14 04:32:46 | | | | |
| 4 | André T. S. Vicente | Meta information modification | 3180 | 2022-10-15 15:02:31 | | |
Video Upload Options
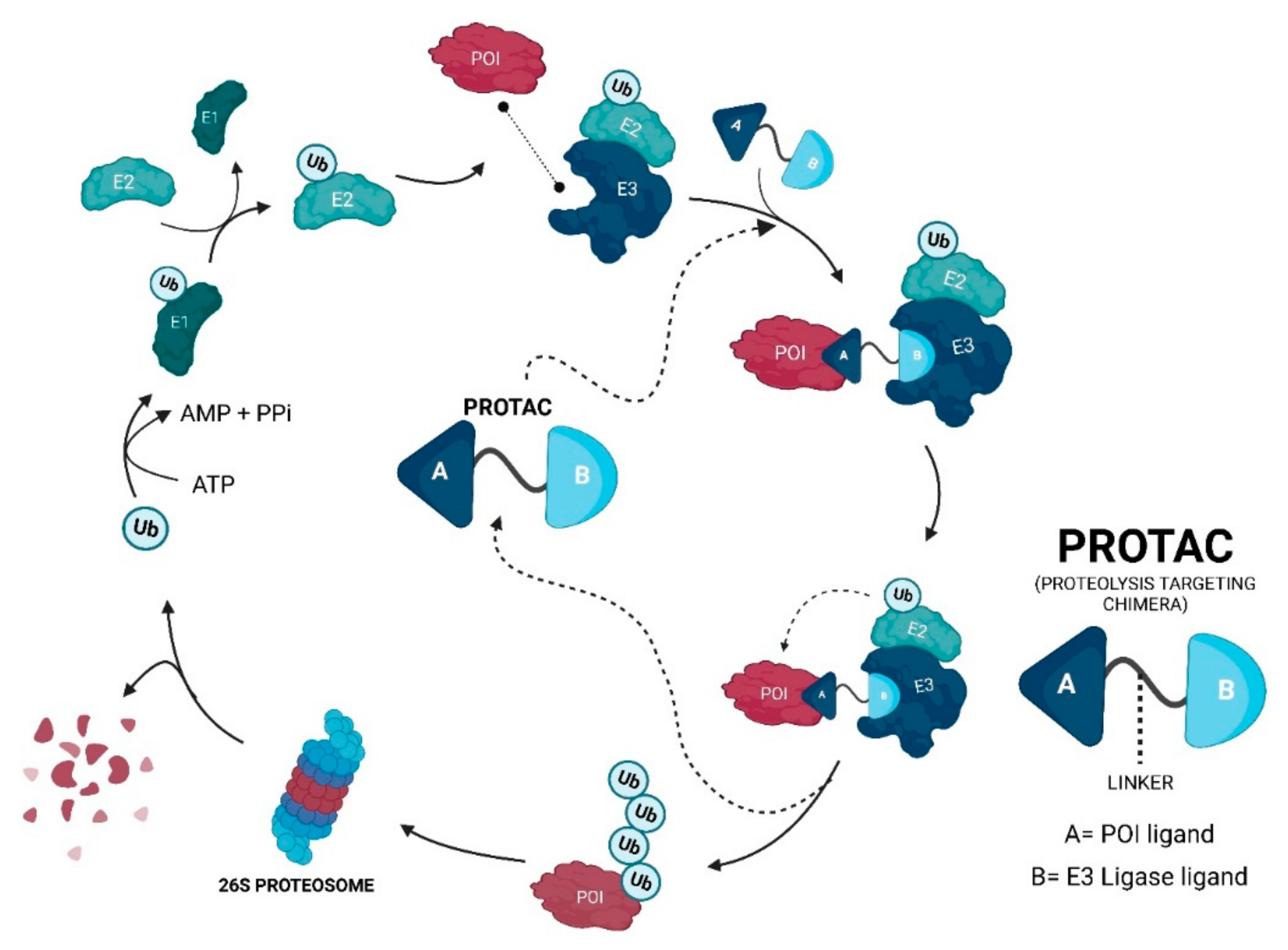
Proteolysis-targeting chimeras (PROTACs) are molecules that selectively degrade a protein of interest (POI). The incorporation of ligands that recruit mouse double minute 2 (MDM2) into PROTACs, forming the so-called MDM2-based PROTACs, has shown promise in cancer treatment due to its dual mechanism of action: a PROTAC that recruits MDM2 prevents its binding to p53, resulting not only in the degradation of POI but also in the increase of intracellular levels of the p53 suppressor, with the activation of a whole set of biological processes, such as cell cycle arrest or apoptosis. In addition, these PROTACs, in certain cases, allow for the degradation of the target, with nanomolar potency, in a rapid and sustained manner over time, with less susceptibility to the development of resistance and tolerance, without causing changes in protein expression, and with selectivity to the target, including the respective isoforms or mutations, and to the cell type, overcoming some limitations associated with the use of inhibitors for the same therapeutic target.
1. PROteolysis-TArgeting Chimeras—PROTACs
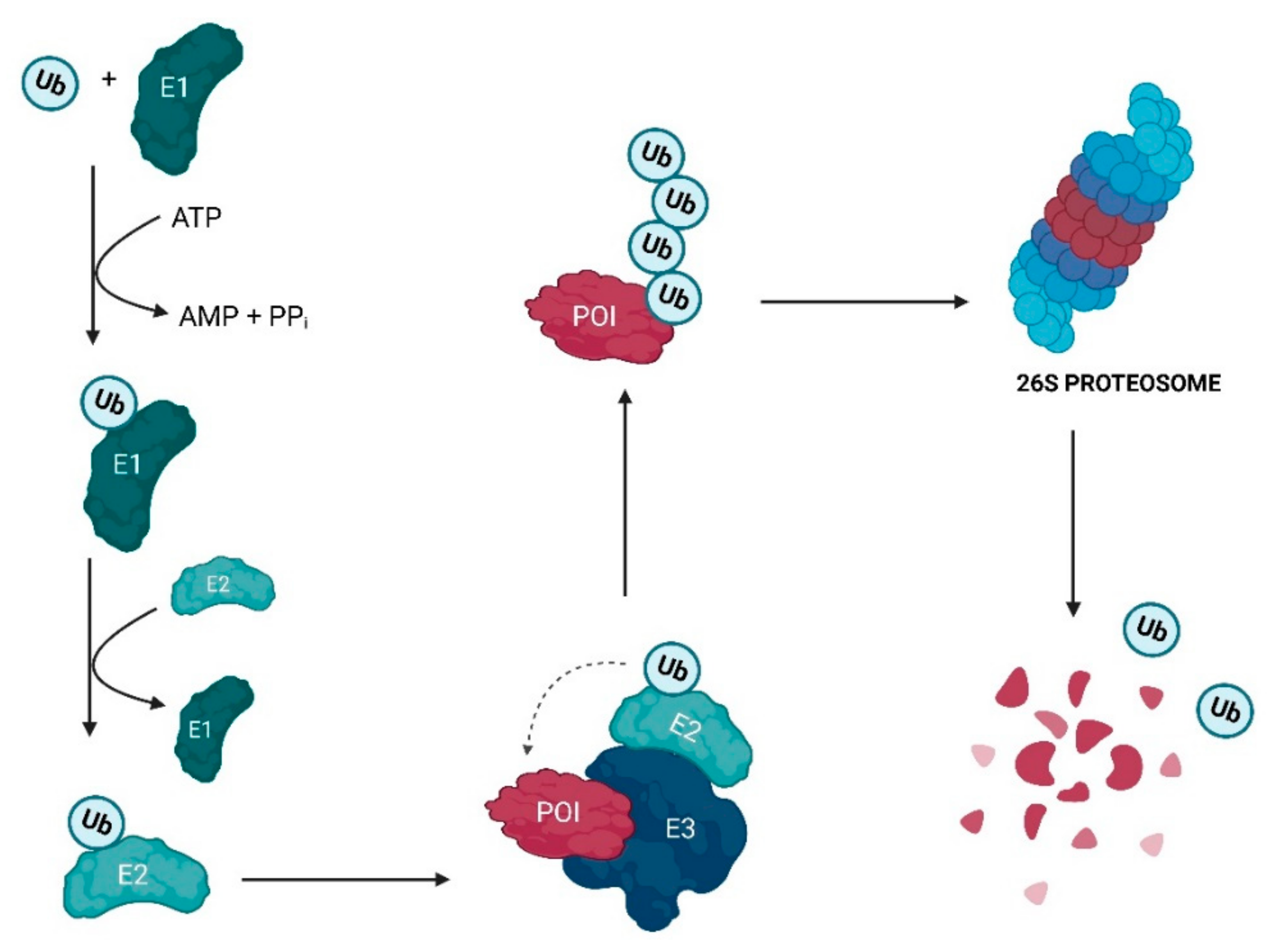
2. Evolutionary Perspective of PROTACs
2.1. First Generation—Peptide-Based PROTACs (2001–2008)
2.2. Second Generation—Small Molecule-Based PROTACs
2.3. Third Generation—Spatiotemporal Controllable PROTACs
3. PROTACs That Recruit MDM2 E3 Ligase
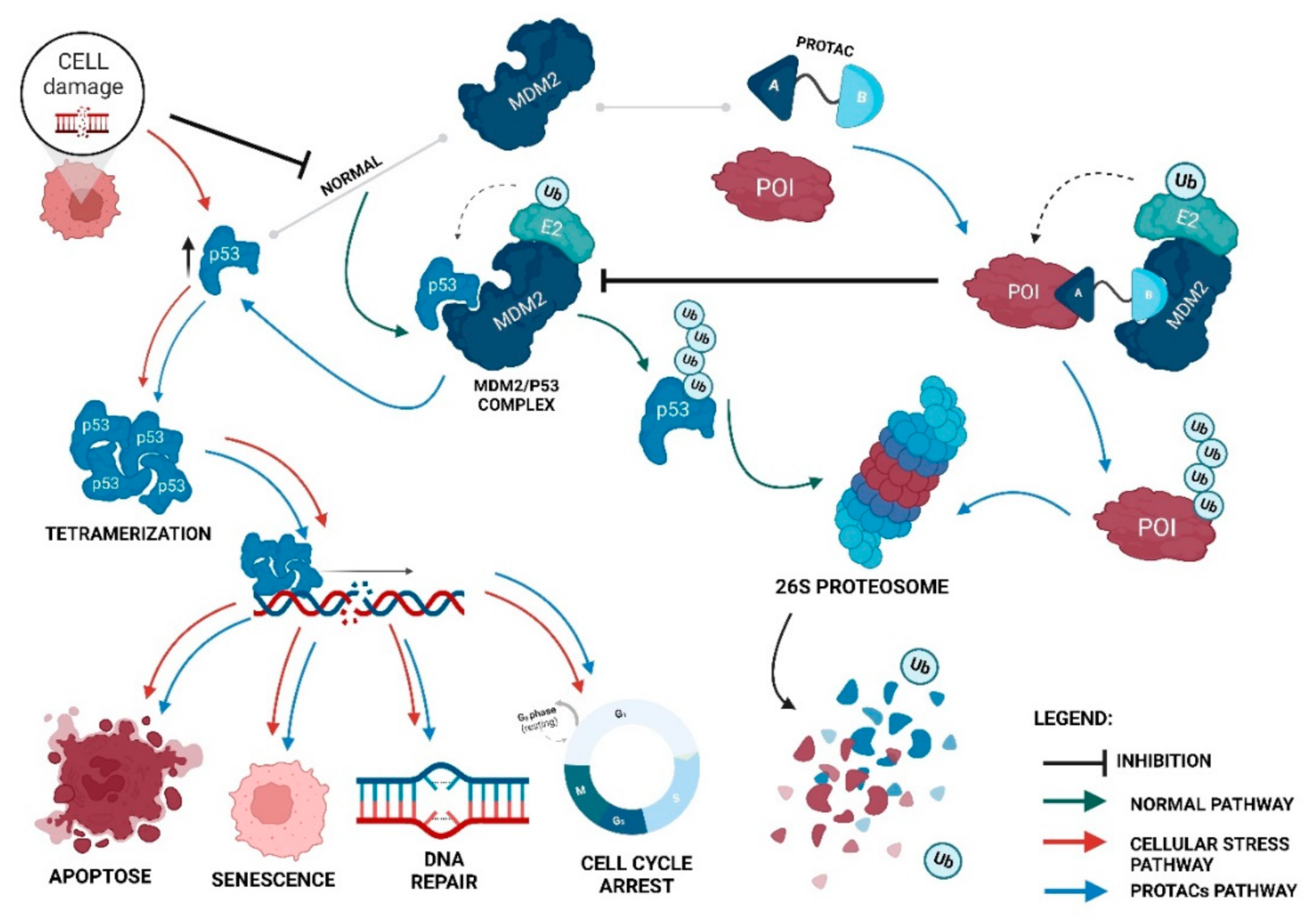
4. Advantages of MDM2-Based PROTACs
-
PROTACs can display cooperativity—binding to a second protein in PROTACs can lead to the formation of the ternary complex being favored (positive cooperativity), or it can lead to the formation of binary complexes (negative cooperativity) or simply remain not affected (neutral cooperativity). If positive cooperativity exists, a stable ternary complex can be formed even in the presence of low affinities with the POI [38][39];
-
PROTACs are less susceptible to the development of resistance—the resistance me-chanism will have to encompass both of the pathways of the PROTAC, given that, in the absence of the degradation capacity, the inhibition of the target or MDM2 by itself may still have anticancer action [42].
5. Disadvantages of MDM2-Based PROTACs
6. Conclusions
References
- Mariell Pettersson; Craig M. Crews; PROteolysis TArgeting Chimeras (PROTACs) — Past, present and future. Drug Discovery Today: Technologies 2019, 31, 15-27, 10.1016/j.ddtec.2019.01.002.
- Kathleen M. Sakamoto; Kyung B. Kim; Akiko Kumagai; Frank Mercurio; Craig M. Crews; Raymond J. Deshaies; Protacs: Chimeric molecules that target proteins to the Skp1–Cullin–F box complex for ubiquitination and degradation. Proceedings of the National Academy of Sciences 2001, 98, 8554-8559, 10.1073/pnas.141230798.
- Yutian Zou; Danhui Ma; Yinyin Wang; The PROTAC technology in drug development. Cell Biochemistry and Function 2019, 37, 21-30, 10.1002/cbf.3369.
- Markella Konstantinidou; Jingyao Li; Bidong Zhang; Zefeng Wang; Shabnam Shaabani; Frans Ter Brake; Khaled Essa; Alexander Dömling; PROTACs– a game-changing technology. Expert Opinion on Drug Discovery 2019, 14, 1255-1268, 10.1080/17460441.2019.1659242.
- Ke Li; Craig M. Crews; PROTACs: past, present and future. Chemical Society Reviews 2022, 51, 5214-5236, 10.1039/d2cs00193d.
- Ashley R. Schneekloth; Mathieu Pucheault; Hyun Seop Tae; Craig M. Crews; Targeted intracellular protein degradation induced by a small molecule: En route to chemical proteomics. Bioorganic & Medicinal Chemistry Letters 2008, 18, 5904-5908, 10.1016/j.bmcl.2008.07.114.
- Brandon Dale; Meng Cheng; Kwang-Su Park; H. Ümit Kaniskan; Yue Xiong; Jian Jin; Advancing targeted protein degradation for cancer therapy. Nature Cancer 2021, 21, 638-654, 10.1038/s41568-021-00365-x.
- Gary Kleiger; Thibault Mayor; Perilous journey: a tour of the ubiquitin–proteasome system. Trends in Cell Biology 2014, 24, 352-359, 10.1016/j.tcb.2013.12.003.
- Markella Konstantinidou; Jingyao Li; Bidong Zhang; Zefeng Wang; Shabnam Shaabani; Frans Ter Brake; Khaled Essa; Alexander Dömling; PROTACs– a game-changing technology. Expert Opinion on Drug Discovery 2019, 14, 1255-1268, 10.1080/17460441.2019.1659242.
- Deepa Nath; Sadaf Shadan; The ubiquitin system. Nature Cell Biology 2009, 458, 421-421, 10.1038/458421a.
- Mohammed A. Mansour; Ubiquitination: Friend and foe in cancer. The International Journal of Biochemistry & Cell Biology 2018, 101, 80-93, 10.1016/j.biocel.2018.06.001.
- Markella Konstantinidou; Jingyao Li; Bidong Zhang; Zefeng Wang; Shabnam Shaabani; Frans Ter Brake; Khaled Essa; Alexander Dömling; PROTACs– a game-changing technology. Expert Opinion on Drug Discovery 2019, 14, 1255-1268, 10.1080/17460441.2019.1659242.
- Zhenyi Hu; Craig M. Crews; Recent Developments in PROTAC‐Mediated Protein Degradation: From Bench to Clinic. ChemBioChem 2021, 23, 1-23, 10.1002/cbic.202100270.
- Chaoguo Cao; Ming He; Liguo Wang; Yuna He; Yu Rao; Chemistries of bifunctional PROTAC degraders. Chemical Society Reviews 2022, 51, 7066–7114, 10.1039/d2cs00220e.
- Markella Konstantinidou; Jingyao Li; Bidong Zhang; Zefeng Wang; Shabnam Shaabani; Frans Ter Brake; Khaled Essa; Alexander Dömling; PROTACs– a game-changing technology. Expert Opinion on Drug Discovery 2019, 14, 1255-1268, 10.1080/17460441.2019.1659242.
- Jing Liu; Jia Ma; Yi Liu; Jun Xia; Yuyun Li; Z. Peter Wang; Wenyi Wei; PROTACs: A novel strategy for cancer therapy. Seminars in Cancer Biology 2020, 67, 171-179, 10.1016/j.semcancer.2020.02.006.
- Xin Zhou; Ru Dong; Jin-Yang Zhang; Xin Zheng; Li-Ping Sun; PROTAC: A promising technology for cancer treatment. European Journal of Medicinal Chemistry 2020, 203, 112539, 10.1016/j.ejmech.2020.112539.
- Kathleen M. Sakamoto; Kyung B. Kim; Akiko Kumagai; Frank Mercurio; Craig M. Crews; Raymond J. Deshaies; Protacs: Chimeric molecules that target proteins to the Skp1–Cullin–F box complex for ubiquitination and degradation. Proceedings of the National Academy of Sciences 2001, 98, 8554-8559, 10.1073/pnas.141230798.
- Kathleen M. Sakamoto; Kyung B. Kim; Rati Verma; Andy Ransick; Bernd Stein; Craig M. Crews; Raymond Deshaies; Development of Protacs to Target Cancer-promoting Proteins for Ubiquitination and Degradation. Molecular & Cellular Proteomics 2003, 2, 1350-1358, 10.1074/mcp.t300009-mcp200.
- John S. Schneekloth Jr.; Fabiana N. Fonseca; Michael Koldobskiy; Amit Mandal; Raymond Deshaies; Kathleen Sakamoto; Craig M. Crews; Chemical Genetic Control of Protein Levels: Selective in Vivo Targeted Degradation. Journal of the American Chemical Society 2004, 126, 3748-3754, 10.1021/ja039025z.
- A A Rodriguez-Gonzalez; K Cyrus; M Salcius; K Kim; C M Crews; R J Deshaies; K M Sakamoto; Targeting steroid hormone receptors for ubiquitination and degradation in breast and prostate cancer. Oncogene 2008, 27, 7201-7211, 10.1038/onc.2008.320.
- Yukihiro Itoh; Minoru Ishikawa; Mikihiko Naito; Yuichi Hashimoto; Protein Knockdown Using Methyl Bestatin−Ligand Hybrid Molecules: Design and Synthesis of Inducers of Ubiquitination-Mediated Degradation of Cellular Retinoic Acid-Binding Proteins. Journal of the American Chemical Society 2010, 132, 5820-5826, 10.1021/ja100691p.
- Georg E. Winter; Dennis L. Buckley; Joshiawa Paulk; Justin M. Roberts; Amanda Souza; Sirano Dhe-Paganon; James E. Bradner; Phthalimide conjugation as a strategy for in vivo target protein degradation. Science 2015, 348, 1376-1381, 10.1126/science.aab1433.
- Dennis L. Buckley; Jeffrey L. Gustafson; Inge Van Molle; Anke G. Roth; Hyun Seop Tae; Peter C. Gareiss; William L. Jorgensen; Alessio Ciulli; Craig M. Crews; Small-Molecule Inhibitors of the Interaction between the E3 Ligase VHL and HIF1α. Angewandte Chemie International Edition 2012, 51, 11463-11467, 10.1002/anie.201206231.
- Zhenyi Hu; Craig M. Crews; Recent Developments in PROTAC‐Mediated Protein Degradation: From Bench to Clinic. ChemBioChem 2021, 23, 1–23, 10.1002/cbic.202100270.
- John Hines; Jonathan D. Gough; Timothy W. Corson; Craig M. Crews; Posttranslational protein knockdown coupled to receptor tyrosine kinase activation with phosphoPROTACs. Proceedings of the National Academy of Sciences 2013, 110, 8942-8947, 10.1073/pnas.1217206110.
- Jing Liu; He Chen; Leina Ma; Zhixiang He; Dong Wang; Yi Liu; Qian Lin; Tinghu Zhang; Nathanael Gray; H. Ümit Kaniskan; et al.Jian JinWenyi Wei Light-induced control of protein destruction by opto-PROTAC. Science Advances 2020, 6, eaay5154, 10.1126/sciadv.aay5154.
- Martin Reynders; Bryan S. Matsuura; Marleen Bérouti; Daniele Simoneschi; Antonio Marzio; Michele Pagano; Dirk Trauner; PHOTACs enable optical control of protein degradation. Science Advances 2020, 6, 0, 10.1126/sciadv.aay5064.
- Patrick Pfaff; Kusal Samarasinghe; Craig M. Crews; Erick M. Carreira; Reversible Spatiotemporal Control of Induced Protein Degradation by Bistable PhotoPROTACs. ACS Central Science 2019, 5, 1682-1690, 10.1021/acscentsci.9b00713.
- Honorine Lebraud; David J. Wright; Christopher N. Johnson; Tom D. Heightman; Protein Degradation by In-Cell Self-Assembly of Proteolysis Targeting Chimeras. ACS Central Science 2016, 2, 927-934, 10.1021/acscentsci.6b00280.
- Marı́a Maneiro; Nafsika Forte; Maria M. Shchepinova; Cyrille S. Kounde; Vijay Chudasama; James Richard Baker; Edward W. Tate; Antibody–PROTAC Conjugates Enable HER2-Dependent Targeted Protein Degradation of BRD4. ACS Chemical Biology 2020, 15, 1306-1312, 10.1021/acschembio.0c00285.
- Kathleen M. Sakamoto; Kyung B. Kim; Akiko Kumagai; Frank Mercurio; Craig M. Crews; Raymond J. Deshaies; Protacs: Chimeric molecules that target proteins to the Skp1–Cullin–F box complex for ubiquitination and degradation. Proceedings of the National Academy of Sciences 2001, 98, 8554-8559, 10.1073/pnas.141230798.
- Scott D. Edmondson; Bin Yang; Charlene Fallan; Proteolysis targeting chimeras (PROTACs) in ‘beyond rule-of-five’ chemical space: Recent progress and future challenges. Bioorganic & Medicinal Chemistry Letters 2019, 29, 1555-1564, 10.1016/j.bmcl.2019.04.030.
- Abiodun Anifowose; Ayodeji A. Agbowuro; Xiaoxiao Yang; Binghe Wang; Anticancer strategies by upregulating p53 through inhibition of its ubiquitination by MDM2. Medicinal Chemistry Research 2020, 29, 1105-1121, 10.1007/s00044-020-02574-9.
- Shusuke Tomoshige; Minoru Ishikawa; In vivo synthetic chemistry of proteolysis targeting chimeras (PROTACs). Bioorganic & Medicinal Chemistry 2021, 41, 116221, 10.1016/j.bmc.2021.116221.
- Cristina Nieto-Jiménez; Esther Cabañas Morafraile; Carlos Alonso-Moreno; Alberto Ocaña; Clinical considerations for the design of PROTACs in cancer. Molecular Cancer 2022, 21, 1-9, 10.1186/s12943-022-01535-7.
- Sainan An; Liwu Fu; Small-molecule PROTACs: An emerging and promising approach for the development of targeted therapy drugs. eBioMedicine 2018, 36, 553-562, 10.1016/j.ebiom.2018.09.005.
- Gary Kleiger; Thibault Mayor; Perilous journey: a tour of the ubiquitin–proteasome system. Trends in Cell Biology 2014, 24, 352-359, 10.1016/j.tcb.2013.12.003.
- Markella Konstantinidou; Jingyao Li; Bidong Zhang; Zefeng Wang; Shabnam Shaabani; Frans Ter Brake; Khaled Essa; Alexander Dömling; PROTACs– a game-changing technology. Expert Opinion on Drug Discovery 2019, 14, 1255-1268, 10.1080/17460441.2019.1659242.
- Shipeng He; Junhui Ma; Yuxin Fang; Ying Liu; Shanchao Wu; Guoqiang Dong; Wei Wang; Chunquan Sheng; Homo-PROTAC mediated suicide of MDM2 to treat non-small cell lung cancer. Acta Pharmaceutica Sinica B 2020, 11, 1617-1628, 10.1016/j.apsb.2020.11.022.
- Xinyi Li; Wenchen Pu; Qingquan Zheng; Min Ai; Song Chen; Yong Peng; Proteolysis-targeting chimeras (PROTACs) in cancer therapy. Molecular Cancer 2022, 21, 1-30, 10.1186/s12943-021-01434-3.
- John Hines; Schan Lartigue; Hanqing Dong; Yimin Qian; Craig M. Crews; MDM2-Recruiting PROTAC Offers Superior, Synergistic Antiproliferative Activity via Simultaneous Degradation of BRD4 and Stabilization of p53. Cancer Research 2019, 79, 251-262, 10.1158/0008-5472.can-18-2918.
- Bohan Ma; Fan Niu; Xiaoyan Qu; Wangxiao He; Chao Feng; Simeng Wang; Zhenlin Ouyang; Jin Yan; Yurong Wen; Dan Xu; et al.Yongping ShaoPeter X. MaWuyuan Lu A tetrameric protein scaffold as a nano-carrier of antitumor peptides for cancer therapy. Biomaterials 2019, 204, 1-12, 10.1016/j.biomaterials.2019.03.004.
- Markella Konstantinidou; Jingyao Li; Bidong Zhang; Zefeng Wang; Shabnam Shaabani; Frans Ter Brake; Khaled Essa; Alexander Dömling; PROTACs– a game-changing technology. Expert Opinion on Drug Discovery 2019, 14, 1255-1268, 10.1080/17460441.2019.1659242.
- Scott D. Edmondson; Bin Yang; Charlene Fallan; Proteolysis targeting chimeras (PROTACs) in ‘beyond rule-of-five’ chemical space: Recent progress and future challenges. Bioorganic & Medicinal Chemistry Letters 2019, 29, 1555-1564, 10.1016/j.bmcl.2019.04.030.
- Yichao Wan; Chunxing Yan; Han Gao; Tingting Liu; Small-molecule PROTACs: novel agents for cancer therapy. Future Medicinal Chemistry 2020, 12, 915-938, 10.4155/fmc-2019-0340.
- Orit Karni-Schmidt; Maria Lokshin; Carol Prives; The Roles of MDM2 and MDMX in Cancer. Annual Review of Pathology: Mechanisms of Disease 2016, 11, 617-644, 10.1146/annurev-pathol-012414-040349.
- Qiuye Zhao; Tianlong Lan; Shang Su; Yu Rao; Induction of apoptosis in MDA-MB-231 breast cancer cells by a PARP1-targeting PROTAC small molecule. Chemical Communications 2018, 55, 369-372, 10.1039/c8cc07813k.

