Activated PXRs play a role in many metabolic pathways, including bile acid metabolism, lipids, and glucose homeostasis, as well as in inflammation
[25]. Recently, their potential role in tumor growth, aggressiveness, relapse, and cancer drug resistance was also noted in mouse tumor xenografts for colon cancer
[26][27][28][29][30]. These various physiological functions make PXRs a potentially valuable therapeutic target
[25][31]. Numerous synthetic or natural ligands, mostly with agonist activities, have been discovered among currently used therapeutics and dietary or natural compounds
[32][33]. Activation of PXRs using their ligands has been connected with many metabolic side effects (e.g., hypercholesterolemia or liver steatosis) as well as with intestinal cancer
[10][25][26][34][35][36]. Therefore, PXR antagonists could have a significant therapeutic value, although designing a novel PXR antagonist is challenging due to the promiscuous nature of the PXR LBP
[37][38][39][40][41][42].
2. The Orthosteric Ligand-Binding Pocket of the PXR
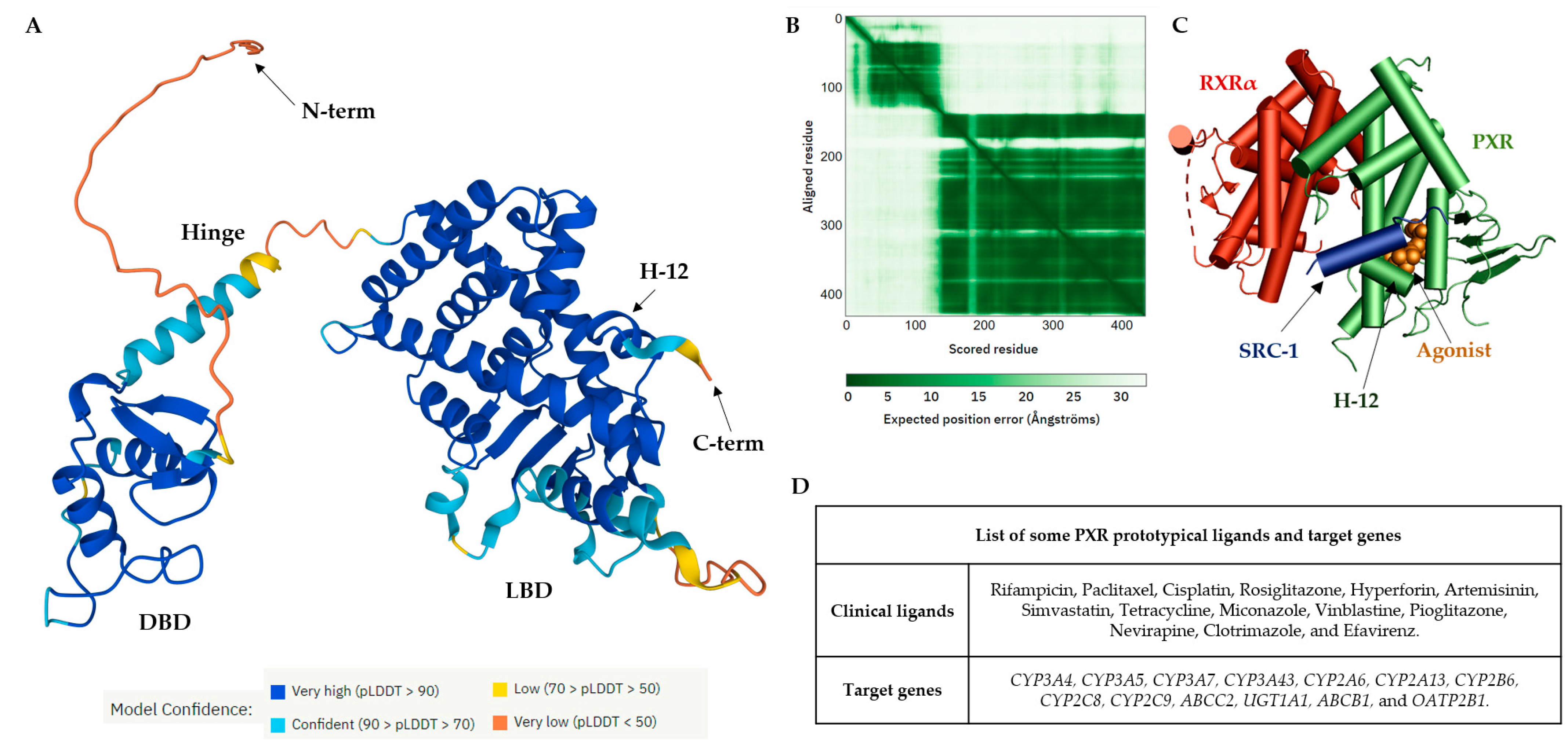
The PXR shares common structural characteristics with other nuclear receptors
[43]. The orthosteric binding site of the PXR receptor is large (>1600 Å
3), dynamic, and flexible enough to bind bulky ligands
[40]. The PXR LBD is characterized by an alpha-helical sandwich structure with a specialized five-stranded beta sheet
[44]. The LBP is deeply embedded in the PXR LBD (
Figure 2A)
[45]. The orthosteric PXR LBP is formed by the helices α3/5/6/7/10/12 (
Figure 2B)
[46]. The PXR ligand-binding cavity was detected at the bottom of the LBD, with the ligand entrance located between the alpha 2 and 6 helices (
Figure 2B)
[40][44]. The PXR was observed as a homodimer in solution, and the crystal structure showed linking with β1′ strands and with support by six intermolecular hydrogen bonds in the monomer
[43].
The PXR LBD shares structural similarities with the VDR and CAR, with a sequence homology of 49.4% and 48.6%, respectively. In humans, the main isoform of the PXR (NM_003889.4, variant 1) consists of 434 amino acids, and the main hydrophobic hot spots are the amino acid residues F288, W299, and Y306, which interact with all reported co-crystallized PXR ligands
[40]. All available 49 PXR LBD crystal structures represent the PXR receptor in its active state and are available for coactivator interactions through helix 12 (H12) for the following transcriptional activation. Due to the destabilization of the PXR LBD, to date no crystal structure of a PXR antagonist has been reported. Therefore, as an alternative to crystallography, the biophysical technique of hydrogen-deuterium exchange mass spectrometry (HDX-MS) was used to analyze the PXR–antagonist interactions
[41].
The structural characteristics of PXR ligands are determined by the nature of the PXR binding pocket: (i) the PXR LBP is large and highly hydrophobic; and (ii) the polar residues Ser247, Gln285, His407, and Arg410 define the ligand binding position in the PXR LBP via strong hydrogen bond (H-bond) interactions. Ser247 and Gln285 residues are involved in the orientation of the ligand binding and the His407 and Arg410 side chains are flexible to accommodate various sizes of ligands in the LBP
[47].
Recently, a detailed examination of the PXR antagonist/inverse agonist SPA70 binding has been conducted. The distinction of the PXR antagonist/inverse agonist (SPA70) from agonist (SJB7) binding showed that the agonist should have the hydrogen bond to the polar residue of Ser247 in helix 10 as well as proximity to the helix 12 region. In contrast, the antagonist has interaction with Ser247 residue and/or has weak contacts with helix 12
[39]. Since the PXR agonist SJB7 and the antagonist SPA70 bind to the same ligand-binding pocket in the PXR in the combined agonist and antagonist conformation, it is difficult to discriminate agonist and antagonist binding sites in the PXR LBD
[39][41]. Notably, a single PXR LBP mutation (W299A) converts the activity of SPA70 from inhibition to activation
[48].
It has recently been reported that the designed PXR molecular glue SJPYT-195, which is composed of SPA70 linked with the CRBN ligand (thalidomide), degraded the GSPT1 translation termination factor instead of the PXR
[49], but the loss of GSPT1 decreased the level of PXR protein in human colon cancer cells (SNU-C4)
[49]. SJPYT-195 weakly bound the PXR LBD, suggesting that long-length linkers may be more favorable in the design of potent PROTACs for PXR
[49][50]. Researchers can also speculate that PXR allosteric sites could be another strategy for the successful targeting of PXR protein degradation (
Figure 2)
[51].
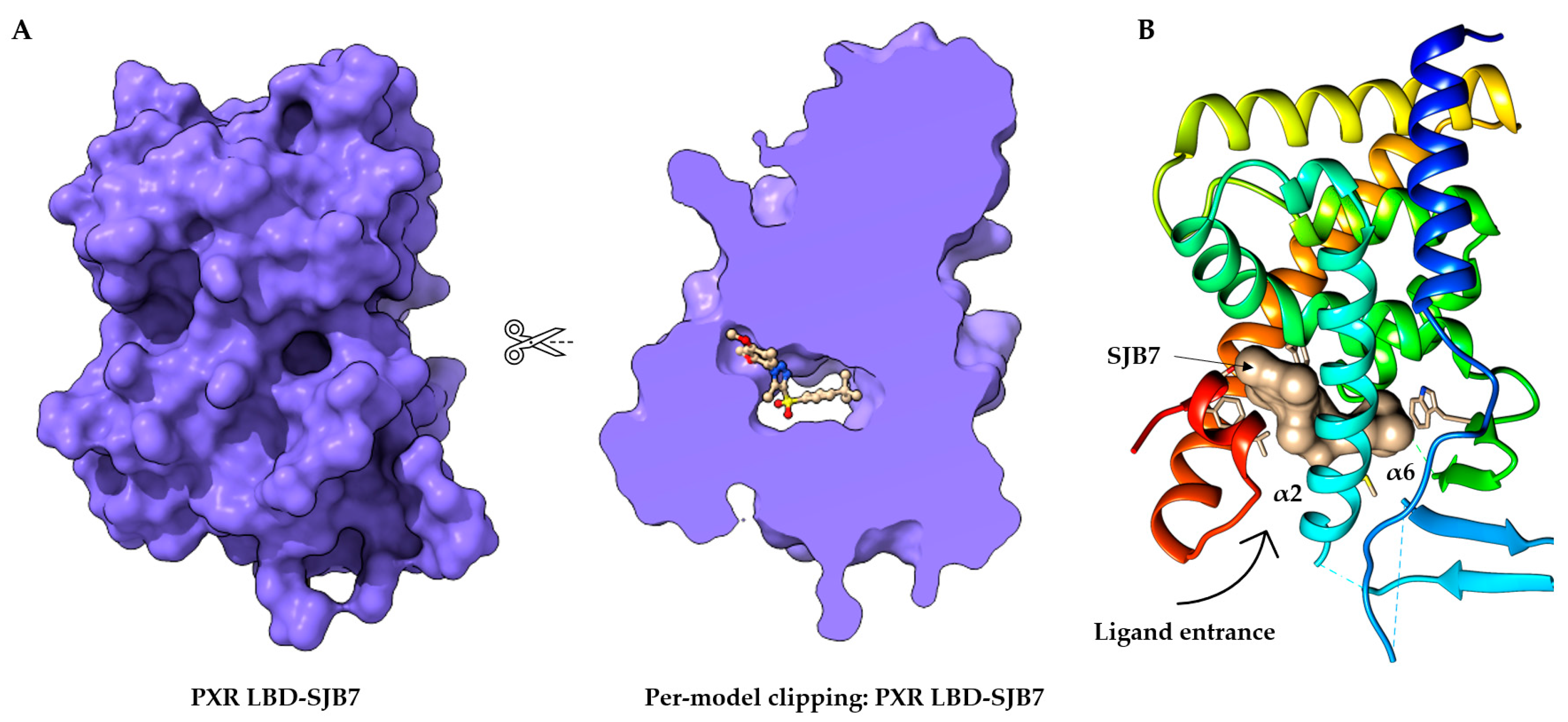
Figure 2. Structure of the orthosteric PXR ligand binding site. (
A) Per-model clipping of the PXR ligand binding domain (to visualize invisible portions of a model). The PXR in PDB entry 5X0R has an SJB7 agonist ligand in an interior pocket, visualized using UCSF ChimeraX
[41][52]. (
B) The crystal structure of the PXR LBD complexed with SJB7. Cartoon representation of PXR- SJB7 binding in the orthosteric site. The PXR agonist SJB7 is indicated in brown, with the arrow indicating the entrance of the ligand. All structures are shown in the same orientation
[41][44].
3. Allosteric Modulation of Nuclear Receptors
Allosteric modulators are usually structurally different from orthosteric ligands, and they bind to distinct sites that are spatially distant from orthosteric sites to modulate the activities of orthosteric ligands
[53].
The essential features of a receptor allosteric modulation are: (1) orthosteric and allosteric sites not overlapping, i.e., there is no mutual biomolecular interaction in the binding; (2) the allosteric binding of one ligand to its site can affect the binding of the second ligand to the orthosteric site and vice versa; and (3) the effect of allosteric modulation can be positive or negative depending on the existing orthosteric ligand. This phenomenon is known as “probe dependence”
[54][55]. Allosteric modulation may be inhibited due to protein–protein interactions (e.g., nuclear receptor–coactivator) or a co-bound receptor, such as in the case of the nuclear receptor homodimerization and heterodimerizations as was reported for G protein-coupled receptors
[56].
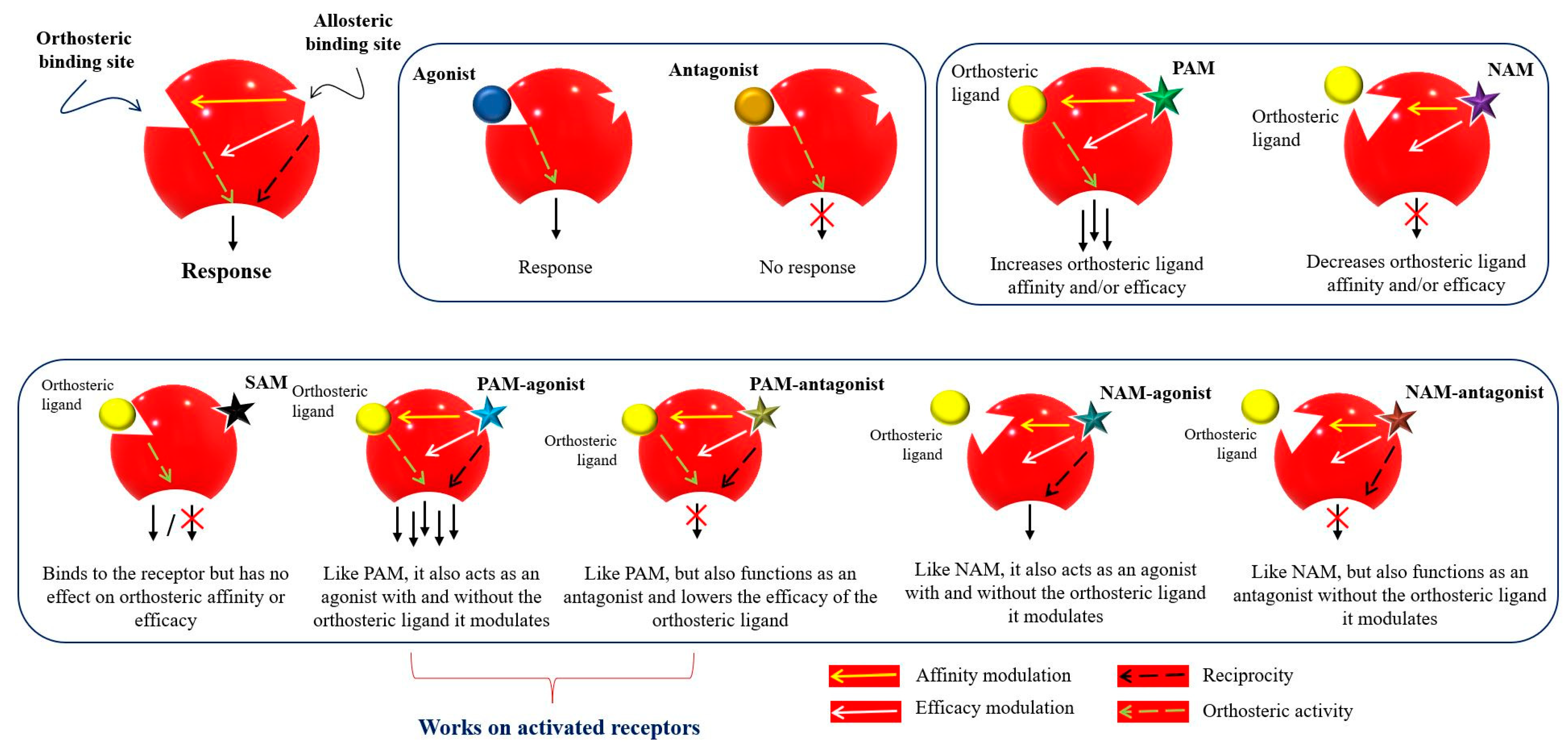
Figure 3 represents the classification of the allosteric modulators and their allosteric properties: affinity modulation, efficacy modulation, reciprocity, and ceiling effect. Affinity modulation indicates the change in the structural conformation of an orthosteric LBP such that the binding affinity of an orthosteric ligand increases or decreases
[57]. Efficacy modulation denotes the increase/decrease in intracellular responses (intrinsic efficacy), depending on the orthosteric ligands (agonist or antagonist)
[57]. The ceiling effect or the saturability effect means that allosteric modulators are non-competitive and maintain a certain saturation level at a certain concentration
[58]. Allosteric modulators may display the possibility of an absolute subtype selectivity for a target protein. For example, a 1700-fold selective allosteric inhibition for phospholipase D1 (PLD1) compared to its subtype PLD2 has been reported
[59]. Allosteric modulators may improve the efficacy and potency of an agonist (e.g., from 2 to 100-fold) in a receptor or may have a synergistic effect
[60][61]. Furthermore, reciprocity or allosteric activation may occur
[54], which means that the receptor is activated directly, without the presence of an orthosteric ligand
[62].
Figure 3. Mode of action and classification of allosteric modulators. Graphical representation of orthosteric and allosteric sites of a receptor. Allosteric ligands bind to a topographically distinct site on a receptor to modulate orthosteric ligand affinity (yellow arrow) and/or efficacy (white arrow). Some allosteric ligands can directly activate their signaling (black arrow). Orthosteric ligands (agonist or antagonist) are colored yellow. Star-shaped allosteric modulators are presented with different colors. PAM—positive allosteric modulator; NAM—negative allosteric modulator; SAM—silent allosteric modulator
[53][57][63][64].
Currently, 202 allosteric modulators have been reported for nuclear receptors
[65], with the following existing allosteric ligand binding surfaces: (i) the AF-2 site; (ii) the binding function 3 (BF-3 site); (iii) the ligand-binding pocket (synergistic); (iv) zinc fingers and response elements; and (v) the AF-1 site
[66][67]. Allosteric modulators have been demonstrated for numerous nuclear receptors; for example, Gabler et al. reported imatinib as a first-in-class allosteric farnesoid X receptor (FXR) modulator that enhances agonist-induced FXR activation in a reporter gene expression assay. The imatinib analogues-16 (I-16) possess extraordinary efficacy (EC
50 = 1.9 nm) and high selectivity over other nuclear receptors
[68][69].
3.1. Allosteric Targeting of the Pregnane X Receptor (PXR)
3.1.1. Duplex—Synergistic Activation of the PXR LBD
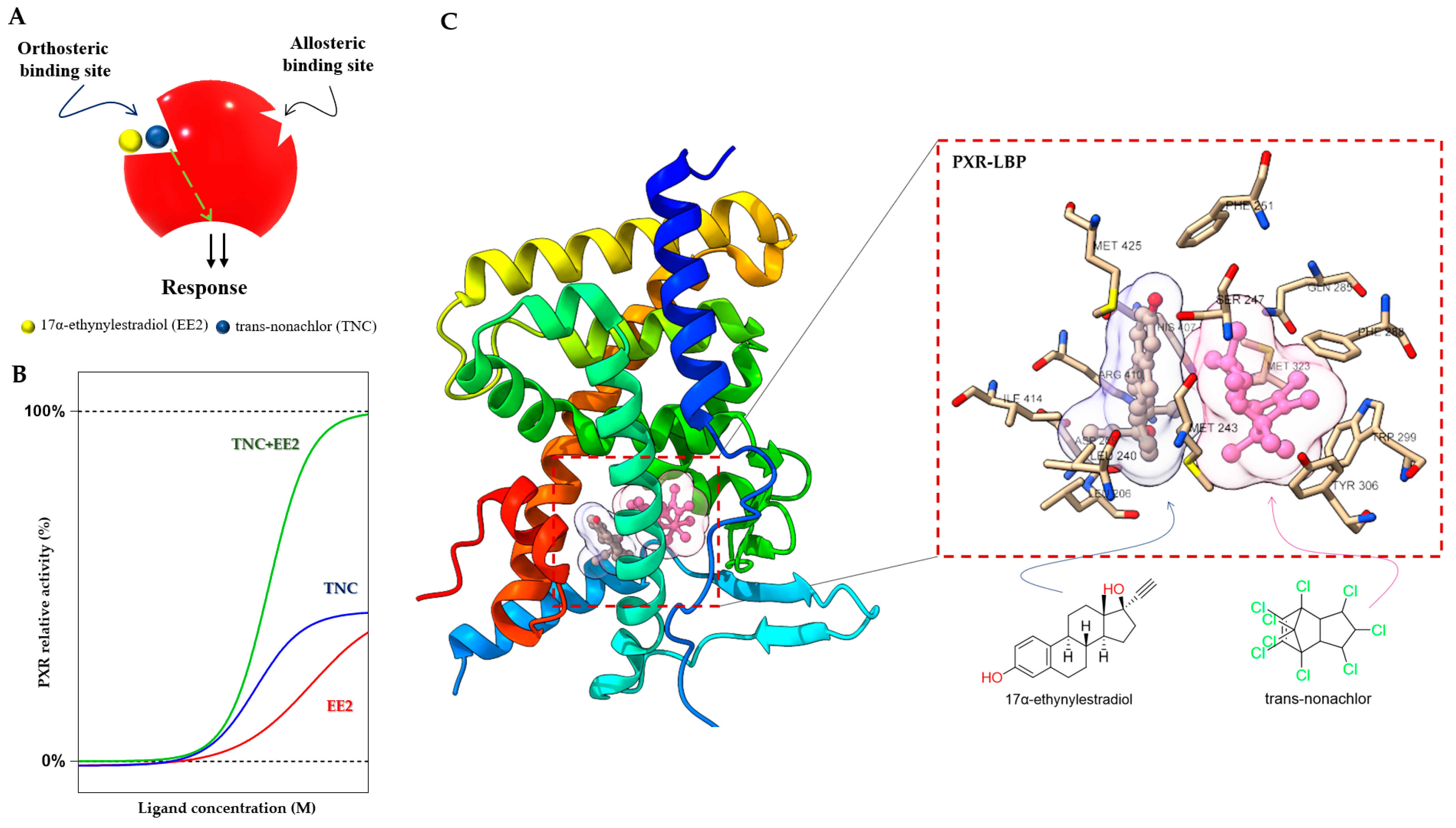
The PXR LBD is able to bind two synthetic ligands concomitantly to the orthosteric ligand binding site (
Figure 4A)
[70], a phenomenon which has been described previously for the PPARγ and ERβ nuclear receptors
[71][72]. Two or more compounds accommodate the same binding sites in the receptors and occupy a space within the canonical LBP near the helix 3 region, leading to enhanced coactivator binding, transactivation, and target gene expression
[70][71][72].
Figure 4. Duplex—synergistic activation of the PXR LBD. (
A) Graphical representation of the duplex model; two ligands (EE2 and TNC) bind to the orthosteric ligand binding site on a receptor to increase the efficacy. (
B) The dose response simulation shows that the combination of TNC and EE2 produces synergistic effects on PXR activation. (
C) Crystal structure of the PXR LBD in complex with EE2 and TNC (PDB-4 × 1 G) and their location highlighted in the ligand binding pocket with interacting amino acid residues
[70][73].
In the case of the PXR, it was found that binary cocktails of the pesticide
trans-nonachlor (TNC) as well as 17α-ethinylestradiol (EE2), the active component of contraceptive pills, produce synergistic activation of the PXR and increase expression of its target
CYP3A4 gene. Interestingly, the single chemicals (EE2 or TNC) were not observed to express the PXR target gene
CYP3A4 in the human hepatocyte
[70]. Synergistic activation of the PXR by EE2 and TNC reached full agonist activity compared to the potent agonist SR12813, but the compounds act as weak agonists when used separately (
Figure 4B).
Small molecules (<500 Da) bind and fill a limited portion of the PXR LBP, leaving empty volume available to accommodate a second compound
[73]. This concept has been proposed as a “supramolecular ligand” assembled from two or more compounds that interact with each other within the LBP of a receptor. TNC binds to the LBP, and EE2 binds to the closely adjacent helix 12 in the PXR LBP (
Figure 4C). This ectopic site near helix 12 may be considered an allosteric site, with bitopic ligands linking orthosteric and allosteric sites to achieve improved PXR affinity or selectivity
[70][74]. The EE2 binding position is a resisted region of the PXR LBP that leaves a significant portion of the pocket unoccupied and available for additional interactions. Supramolecular ligands of the PXR and their properties have been reported to a very limited extent so far, and researchers can expect many more compounds with this activity
[70].
3.1.2. The AF-2 Binding Site at the PXR LBD and Its Ligands
It was found that ligands binding to the AF-2 function of the PXR alter the interactions between a coregulator peptide and the PXR. The ligand-dependent groove of the AF-2 is made up of helixes 3, 4, 5, and 12, which are mainly hydrophobic regions
[37]. The orthosteric PXR LBP ends with a short helix 12 (H12), which is important for the structural organization of the AF-2 region to recruit transcriptional coregulators. Coregulators play an important functional role in the transduction of PXR signals. Both corepressors and coactivators bind to the AF-2 regions through a short amphipathic helical sequence containing the Leu-Xxx-Xxx-Leu-Leu (LXXLL) motif in coactivators or Ile/Leu-Xxx-Xxx-Ile/Val-Ile motifs in corepressors via an electrostatic interaction
[75][76]. When the binding of a ligand is performed, helix H12 undergoes a significant conformational structural change that alters the overall shape of the AF-2 binding site. The PXR ligands alter the AF-2 site after binding into the PXR LBP, and thus modify the recruitment of coactivators or corepressors, resulting in different agonist or antagonist effects of the ligands
[77].
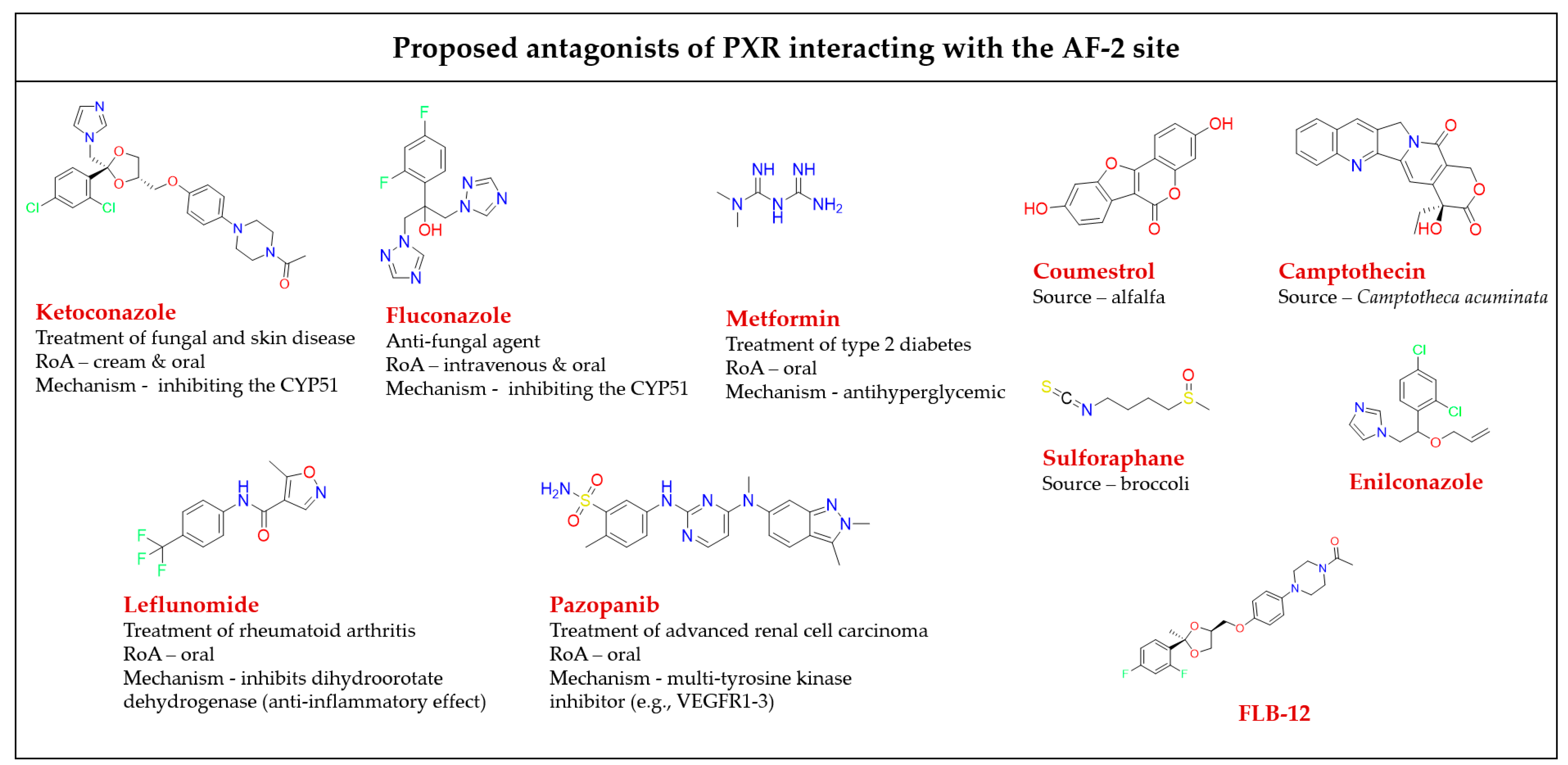
All available PXR antagonist molecules act through similar agonist activation mechanisms, except for some AF-2 disruptors (
Figure 5). The PXR’s ligand binding site is the same for both agonists and antagonists, but there are small residue differences with helix 12
[39][40][78]. In addition, none of the identified AF-2 site binding antagonists possess allosteric properties (selectivity, efficacy, or affinity) (
Table 1)
[54]. Targeting of the AF-2 site always involves issues of nuclear receptor selectivity because the AF-2 site is structurally conserved across subtypes of the nuclear receptor family
[78][79]. For example, ketoconazole, the first identified PXR AF-2 site allosteric modulator and antagonist, is a common inhibitor of activated PXRs, CARs, LXRα/βs, and FXRs
[80][81]. Ketoconazole binding to the surfaces of the AF-2 site suggests that ketoconazole directly blocks a coactivator (e.g., SRC-1) binding, a finding which was confirmed by a double mutant model (T248E/K277Q) in the AF-2 region of the PXR
[81].
The phytoestrogen coumestrol is a natural PXR antagonist proposed to bind to a non-LBP. Biochemical binding assays and LBP-filled mutant (obliterating) studies confirm their surface binding is distinct from the orthosteric site of the LBP
[82]. Additionally, computational pharmacophore and docking analyses showed that the known PXR antagonists coumestrol and sulforaphane also accommodate the AF-2 ligand-binding site
[83].
In another study, the azole compound FLB-12, a derivative of the azole antifungal ketoconazole, antagonized activated PXRs in the hepatocyte cell line and in in vivo models. The triple mutant plasmid of the PXR LBP (S247W/S208W/C284W) was used to confirm its binding sites outside the PXR LBP. FLB-12 disrupts the interaction between the PXR and SRC-1, a finding which was verified by the protein pull-down assay, indicating the location of binding into the AF-2 sites. This antagonist was found to be selective and less toxic as compared to ketoconazole
[84].
Ekins et al. reported residues of the PXR AF-2 ligand binding site. The identified AF-2 ligand binding site is predominantly hydrophobic, and it consists of 15 amino acids (Lys252, Ile255, Lys259, Phe264, Ile269, Glu270, Gln272, Ile273, Ser274, Leu276, Lys277, Pro423, Leu424, Glu427, and Leu428). Lys277 probably serves as a “charge clamp” for the interaction between the coactivator SRC-1 (His687) and the PXR, and it may play a significant role in the initial phase of accommodation of azole molecules into the binding groove of the PXR. Two more azole analogs, enilconazole and fluconazole, have also had interactions confirmed with the AF-2 ligand binding site, as well as antagonist activity shown towards the PXR
[80][85].
Figure 5. Chemical structures of known AF-2 site-binding PXR antagonists. RoA-routes of administration. The chemical structures were drawn in ChemDraw 20.0 software
[80][81][82][83][84][86][87][88][89].
Table 1. A detailed list of antagonists that bind to the PXR AF-2 binding site.
Leflunomide, a drug used clinically for rheumatic arthritis therapy, acts as a PXR antagonist, but as an activator of the CAR
[90]. It was shown that it inhibits the PXR/SRC-1 interaction as demonstrated by site-directed mutagenesis of the AF-2 sites
[83][91].
The pentacyclic alkaloid camptothecin is known as a topoisomerase I inhibitor, and its analogs are approved for colon cancer therapy. Camptothecin was identified as attenuating
CYP3A4 induction by blocking PXR activation via binding outside of the PXR LBP to prevent the recruitment of coactivators (such as SRC-1)
[88].
 +1 credit
+1 credit