The pregnane X receptor (PXR,
NR1I2) is a xenobiotic-activated transcription factor with high levels of expression in the liver. It not only plays a key role in drug metabolism and elimination, but also promotes tumor growth, drug resistance, and metabolic diseases. It has been proposed as a therapeutic target for type II diabetes, metabolic syndrome, and inflammatory bowel disease, and PXR antagonists have recently been considered as a therapy for colon cancer. The identified allosteric sites of the PXR provide new insights into the development of safe and efficient allosteric modulators of the PXR receptor. We therefore propose that novel PXR allosteric sites might be promising targets for treating chronic metabolic diseases and some cancers.
) is a xenobiotic-activated transcription factor with high levels of expression in the liver. It not only plays a key role in drug metabolism and elimination, but also promotes tumor growth, drug resistance, and metabolic diseases. It has been proposed as a therapeutic target for type II diabetes, metabolic syndrome, and inflammatory bowel disease, and PXR antagonists have recently been considered as a therapy for colon cancer. The identified allosteric sites of the PXR provide new insights into the development of safe and efficient allosteric modulators of the PXR receptor. Researchers therefore propose that novel PXR allosteric sites might be promising targets for treating chronic metabolic diseases and some cancers.
- PXR
- pregnane X receptor
- allosteric site
- CAR
- colon cancer
- BF 3
- nuclear receptor
1. Introduction
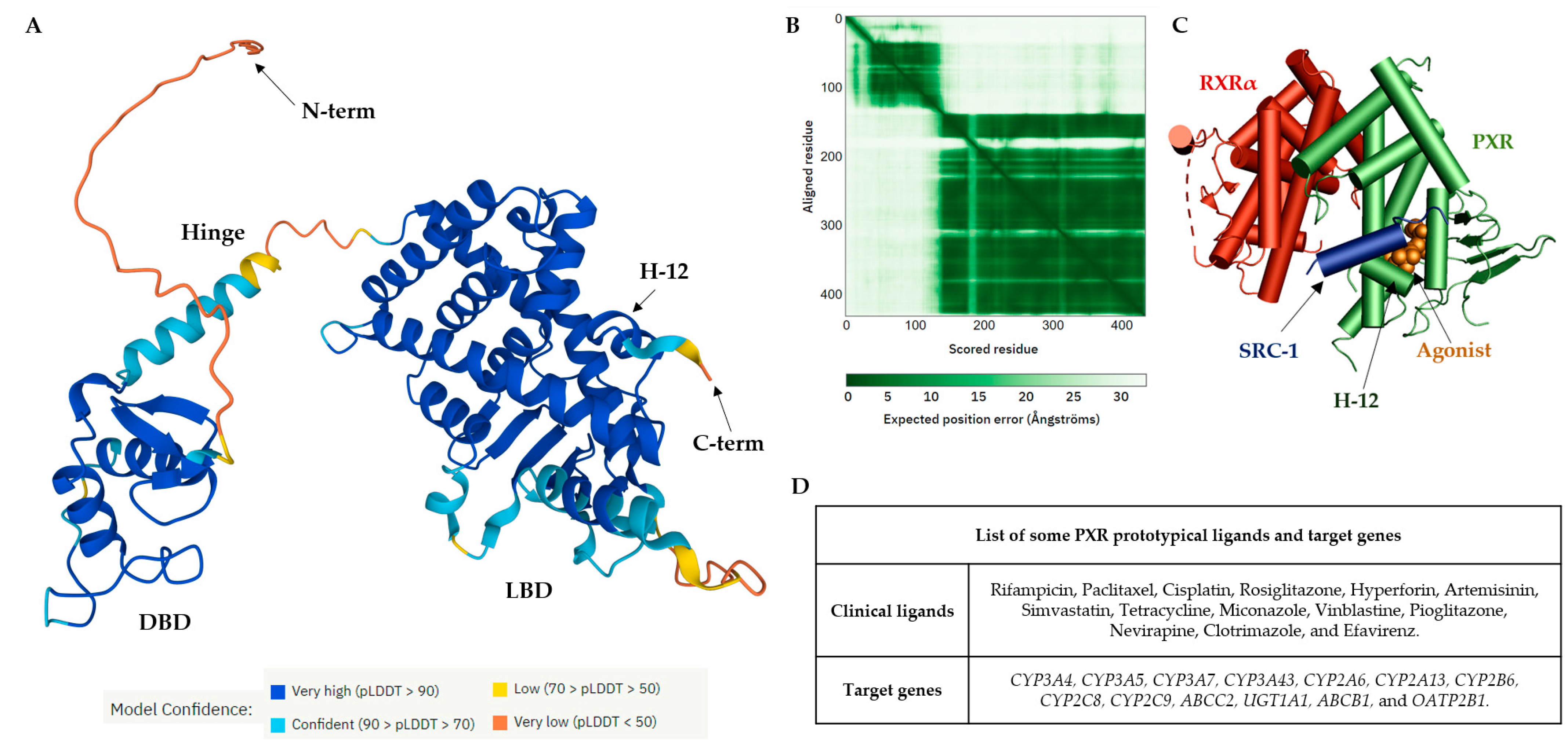
2. The Orthosteric Ligand-Binding Pocket of the PXR
The PXR shares common structural characteristics with other nuclear receptors [43]. The orthosteric binding site of the PXR receptor is large (>1600 Å3), dynamic, and flexible enough to bind bulky ligands [40]. The PXR LBD is characterized by an alpha-helical sandwich structure with a specialized five-stranded beta sheet [44]. The LBP is deeply embedded in the PXR LBD (Figure 2A) [45]. The orthosteric PXR LBP is formed by the helices α3/5/6/7/10/12 (Figure 2B) [46]. The PXR ligand-binding cavity was detected at the bottom of the LBD, with the ligand entrance located between the alpha 2 and 6 helices (Figure 2B) [40,44][40][44]. The PXR was observed as a homodimer in solution, and the crystal structure showed linking with β1′ strands and with support by six intermolecular hydrogen bonds in the monomer [43]. The PXR LBD shares structural similarities with the VDR and CAR, with a sequence homology of 49.4% and 48.6%, respectively. In humans, the main isoform of the PXR (NM_003889.4, variant 1) consists of 434 amino acids, and the main hydrophobic hot spots are the amino acid residues F288, W299, and Y306, which interact with all reported co-crystallized PXR ligands [40]. All available 49 PXR LBD crystal structures represent the PXR receptor in its active state and are available for coactivator interactions through helix 12 (H12) for the following transcriptional activation. Due to the destabilization of the PXR LBD, to date no crystal structure of a PXR antagonist has been reported. Therefore, as an alternative to crystallography, the biophysical technique of hydrogen-deuterium exchange mass spectrometry (HDX-MS) was used to analyze the PXR–antagonist interactions [41]. The structural characteristics of PXR ligands are determined by the nature of the PXR binding pocket: (i) the PXR LBP is large and highly hydrophobic; and (ii) the polar residues Ser247, Gln285, His407, and Arg410 define the ligand binding position in the PXR LBP via strong hydrogen bond (H-bond) interactions. Ser247 and Gln285 residues are involved in the orientation of the ligand binding and the His407 and Arg410 side chains are flexible to accommodate various sizes of ligands in the LBP [47]. Recently, a detailed examination of the PXR antagonist/inverse agonist SPA70 binding has been conducted. The distinction of the PXR antagonist/inverse agonist (SPA70) from agonist (SJB7) binding showed that the agonist should have the hydrogen bond to the polar residue of Ser247 in helix 10 as well as proximity to the helix 12 region. In contrast, the antagonist has interaction with Ser247 residue and/or has weak contacts with helix 12 [39]. Since the PXR agonist SJB7 and the antagonist SPA70 bind to the same ligand-binding pocket in the PXR in the combined agonist and antagonist conformation, it is difficult to discriminate agonist and antagonist binding sites in the PXR LBD [39,41][39][41]. Notably, a single PXR LBP mutation (W299A) converts the activity of SPA70 from inhibition to activation [48]. It has recently been reported that the designed PXR molecular glue SJPYT-195, which is composed of SPA70 linked with the CRBN ligand (thalidomide), degraded the GSPT1 translation termination factor instead of the PXR [49], but the loss of GSPT1 decreased the level of PXR protein in human colon cancer cells (SNU-C4) [49]. SJPYT-195 weakly bound the PXR LBD, suggesting that long-length linkers may be more favorable in the design of potent PROTACs for PXR [49,50][49][50]. WResearchers can also speculate that PXR allosteric sites could be another strategy for the successful targeting of PXR protein degradation (Figure 2) [51].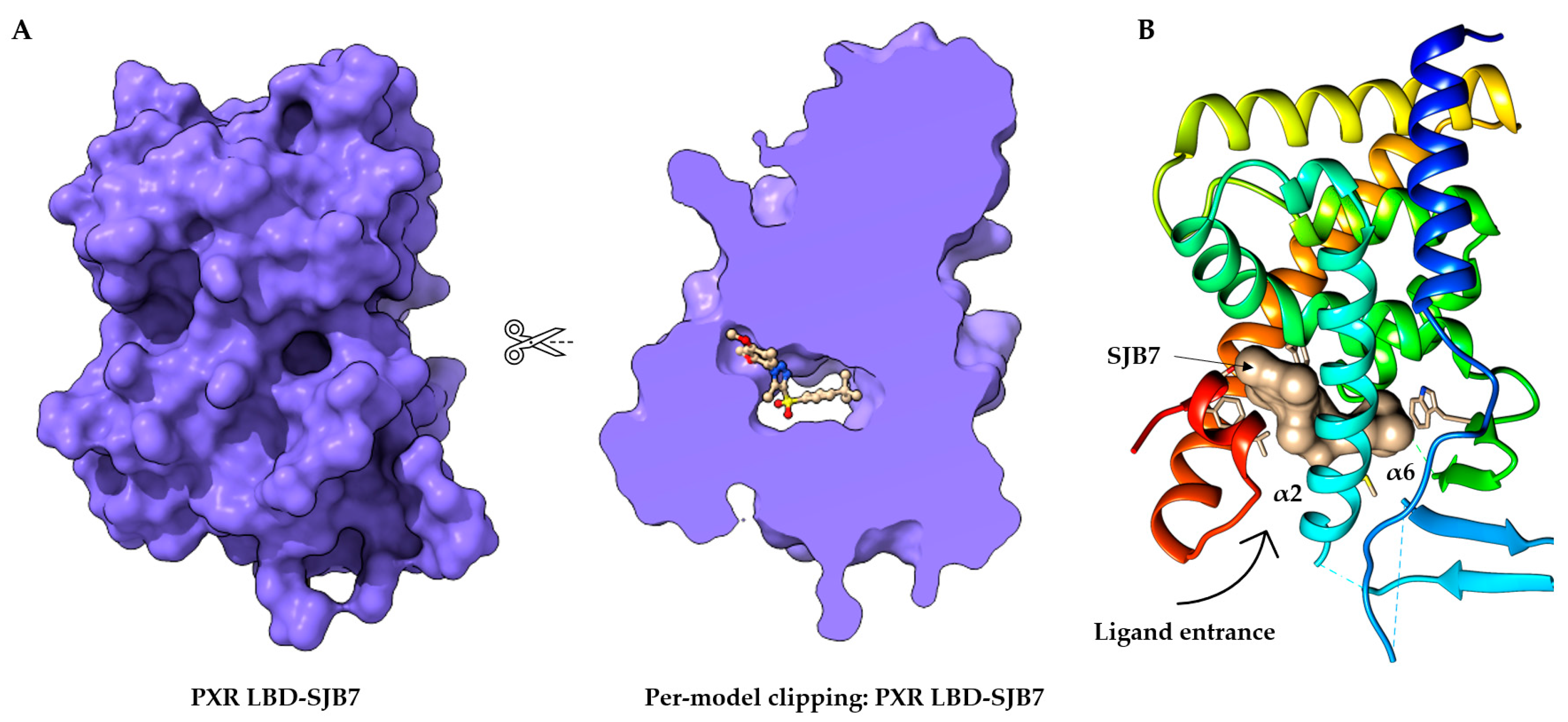
3. Allosteric Modulation of Nuclear Receptors
Allosteric modulators are usually structurally different from orthosteric ligands, and they bind to distinct sites that are spatially distant from orthosteric sites to modulate the activities of orthosteric ligands [53]. The essential features of a receptor allosteric modulation are: (1) orthosteric and allosteric sites not overlapping, i.e., there is no mutual biomolecular interaction in the binding; (2) the allosteric binding of one ligand to its site can affect the binding of the second ligand to the orthosteric site and vice versa; and (3) the effect of allosteric modulation can be positive or negative depending on the existing orthosteric ligand. This phenomenon is known as “probe dependence” [54,55][54][55]. Allosteric modulation may be inhibited due to protein–protein interactions (e.g., nuclear receptor–coactivator) or a co-bound receptor, such as in the case of the nuclear receptor homodimerization and heterodimerizations as was reported for G protein-coupled receptors [56]. Figure 3 represents the classification of the allosteric modulators and their allosteric properties: affinity modulation, efficacy modulation, reciprocity, and ceiling effect. Affinity modulation indicates the change in the structural conformation of an orthosteric LBP such that the binding affinity of an orthosteric ligand increases or decreases [57]. Efficacy modulation denotes the increase/decrease in intracellular responses (intrinsic efficacy), depending on the orthosteric ligands (agonist or antagonist) [57]. The ceiling effect or the saturability effect means that allosteric modulators are non-competitive and maintain a certain saturation level at a certain concentration [58]. Allosteric modulators may display the possibility of an absolute subtype selectivity for a target protein. For example, a 1700-fold selective allosteric inhibition for phospholipase D1 (PLD1) compared to its subtype PLD2 has been reported [59]. Allosteric modulators may improve the efficacy and potency of an agonist (e.g., from 2 to 100-fold) in a receptor or may have a synergistic effect [60,61][60][61]. Furthermore, reciprocity or allosteric activation may occur [54], which means that the receptor is activated directly, without the presence of an orthosteric ligand [62].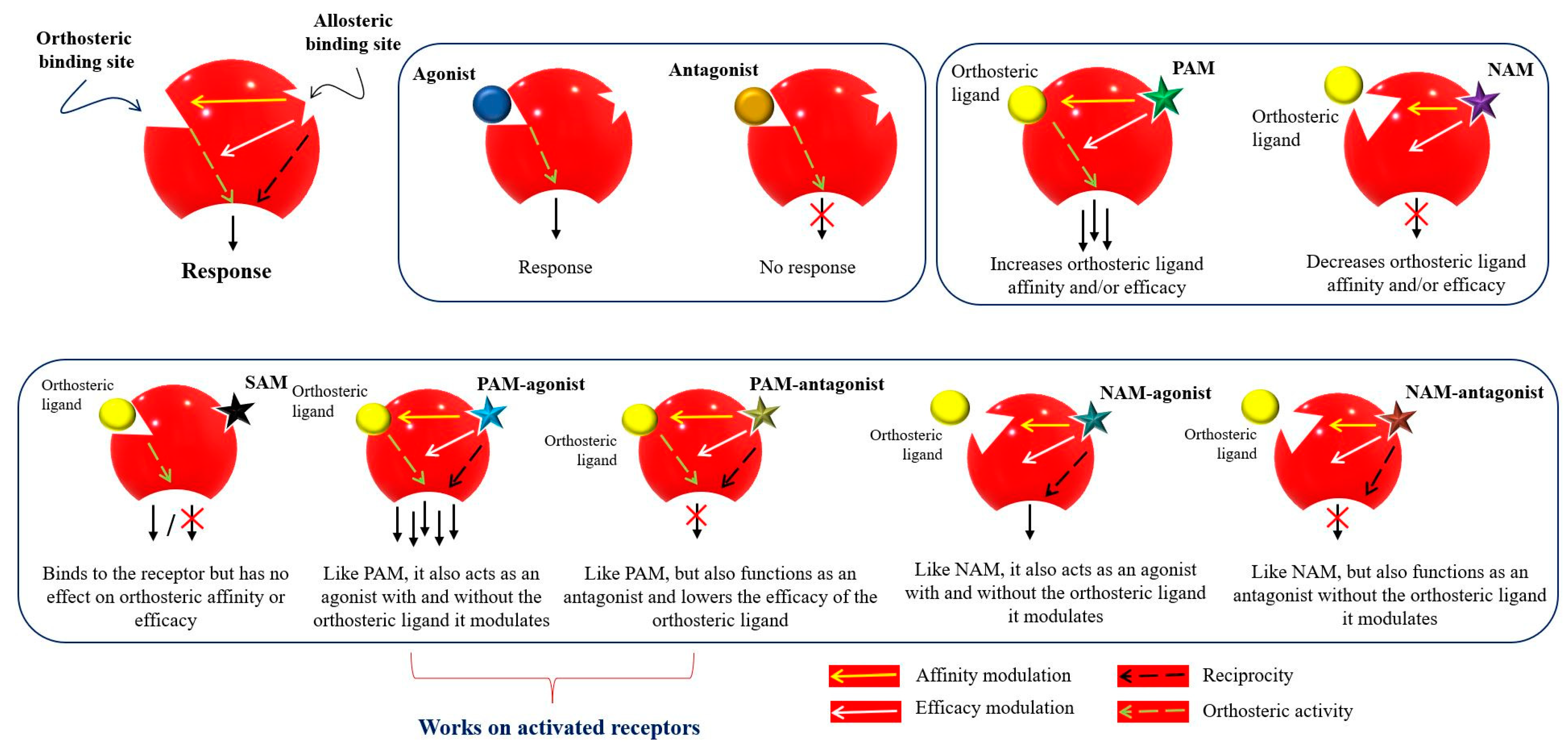
3.1. Allosteric Targeting of the Pregnane X Receptor (PXR)
3.1.1. Duplex—Synergistic Activation of the PXR LBD
The PXR LBD is able to bind two synthetic ligands concomitantly to the orthosteric ligand binding site (Figure 4A) [70], a phenomenon which has been described previously for the PPARγ and ERβ nuclear receptors [71,72][71][72]. Two or more compounds accommodate the same binding sites in the receptors and occupy a space within the canonical LBP near the helix 3 region, leading to enhanced coactivator binding, transactivation, and target gene expression [70,71,72][70][71][72].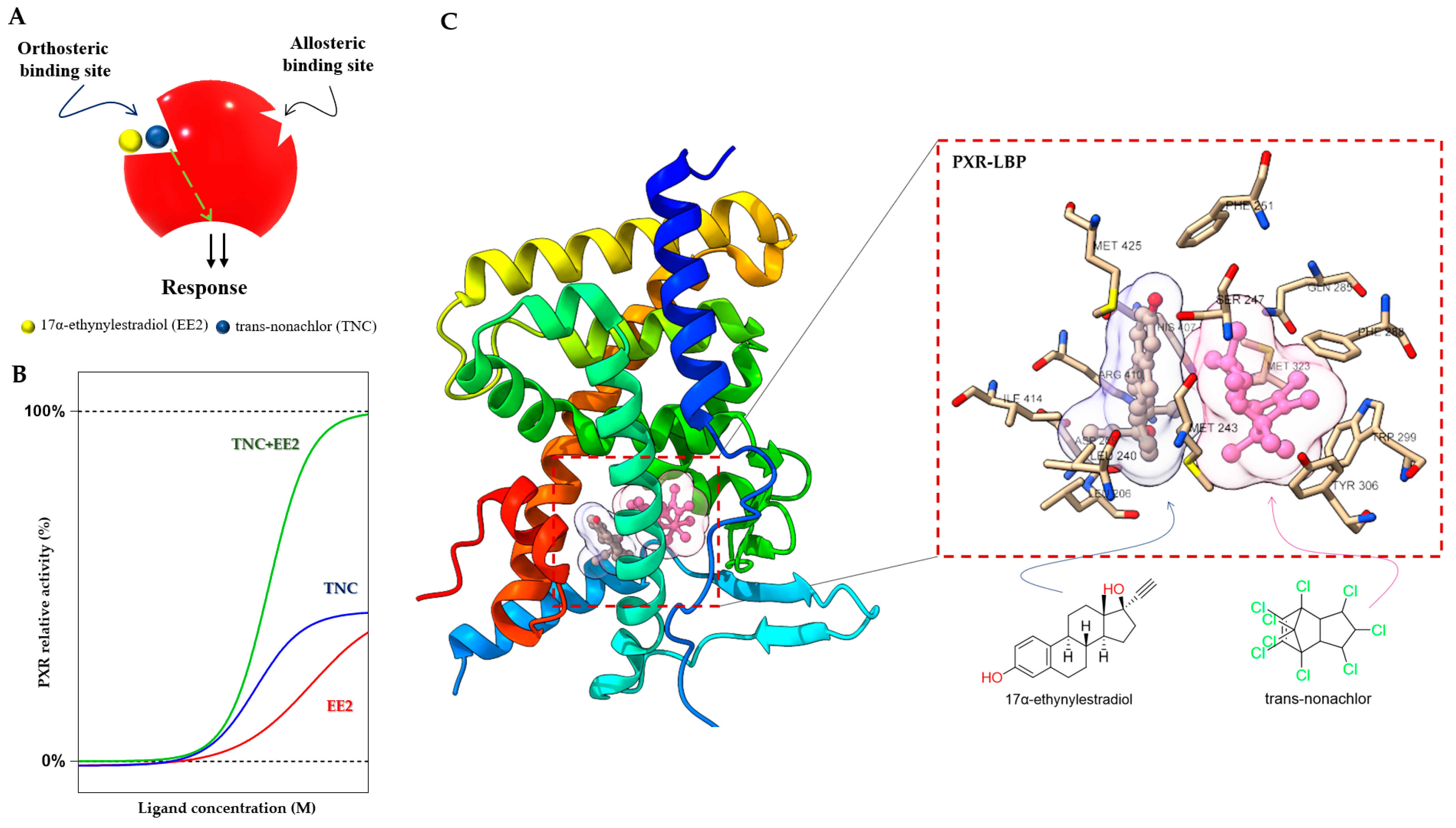
3.1.2. The AF-2 Binding Site at the PXR LBD and Its Ligands
It was found that ligands binding to the AF-2 function of the PXR alter the interactions between a coregulator peptide and the PXR. The ligand-dependent groove of the AF-2 is made up of helixes 3, 4, 5, and 12, which are mainly hydrophobic regions [37]. The orthosteric PXR LBP ends with a short helix 12 (H12), which is important for the structural organization of the AF-2 region to recruit transcriptional coregulators. Coregulators play an important functional role in the transduction of PXR signals. Both corepressors and coactivators bind to the AF-2 regions through a short amphipathic helical sequence containing the Leu-Xxx-Xxx-Leu-Leu (LXXLL) motif in coactivators or Ile/Leu-Xxx-Xxx-Ile/Val-Ile motifs in corepressors via an electrostatic interaction [75,76][75][76]. When the binding of a ligand is performed, helix H12 undergoes a significant conformational structural change that alters the overall shape of the AF-2 binding site. The PXR ligands alter the AF-2 site after binding into the PXR LBP, and thus modify the recruitment of coactivators or corepressors, resulting in different agonist or antagonist effects of the ligands [77]. All available PXR antagonist molecules act through similar agonist activation mechanisms, except for some AF-2 disruptors (Figure 5). The PXR’s ligand binding site is the same for both agonists and antagonists, but there are small residue differences with helix 12 [39,40,78][39][40][78]. In addition, none of the identified AF-2 site binding antagonists possess allosteric properties (selectivity, efficacy, or affinity) (Table 1) [54]. Targeting of the AF-2 site always involves issues of nuclear receptor selectivity because the AF-2 site is structurally conserved across subtypes of the nuclear receptor family [78,79][78][79]. For example, ketoconazole, the first identified PXR AF-2 site allosteric modulator and antagonist, is a common inhibitor of activated PXRs, CARs, LXRα/βs, and FXRs [80,81][80][81]. Ketoconazole binding to the surfaces of the AF-2 site suggests that ketoconazole directly blocks a coactivator (e.g., SRC-1) binding, a finding which was confirmed by a double mutant model (T248E/K277Q) in the AF-2 region of the PXR [81]. The phytoestrogen coumestrol is a natural PXR antagonist proposed to bind to a non-LBP. Biochemical binding assays and LBP-filled mutant (obliterating) studies confirm their surface binding is distinct from the orthosteric site of the LBP [82]. Additionally, computational pharmacophore and docking analyses showed that the known PXR antagonists coumestrol and sulforaphane also accommodate the AF-2 ligand-binding site [83]. In another study, the azole compound FLB-12, a derivative of the azole antifungal ketoconazole, antagonized activated PXRs in the hepatocyte cell line and in in vivo models. The triple mutant plasmid of the PXR LBP (S247W/S208W/C284W) was used to confirm its binding sites outside the PXR LBP. FLB-12 disrupts the interaction between the PXR and SRC-1, a finding which was verified by the protein pull-down assay, indicating the location of binding into the AF-2 sites. This antagonist was found to be selective and less toxic as compared to ketoconazole [84]. Ekins et al. reported residues of the PXR AF-2 ligand binding site. The identified AF-2 ligand binding site is predominantly hydrophobic, and it consists of 15 amino acids (Lys252, Ile255, Lys259, Phe264, Ile269, Glu270, Gln272, Ile273, Ser274, Leu276, Lys277, Pro423, Leu424, Glu427, and Leu428). Lys277 probably serves as a “charge clamp” for the interaction between the coactivator SRC-1 (His687) and the PXR, and it may play a significant role in the initial phase of accommodation of azole molecules into the binding groove of the PXR. Two more azole analogs, enilconazole and fluconazole, have also had interactions confirmed with the AF-2 ligand binding site, as well as antagonist activity shown towards the PXR [80,85][80][85].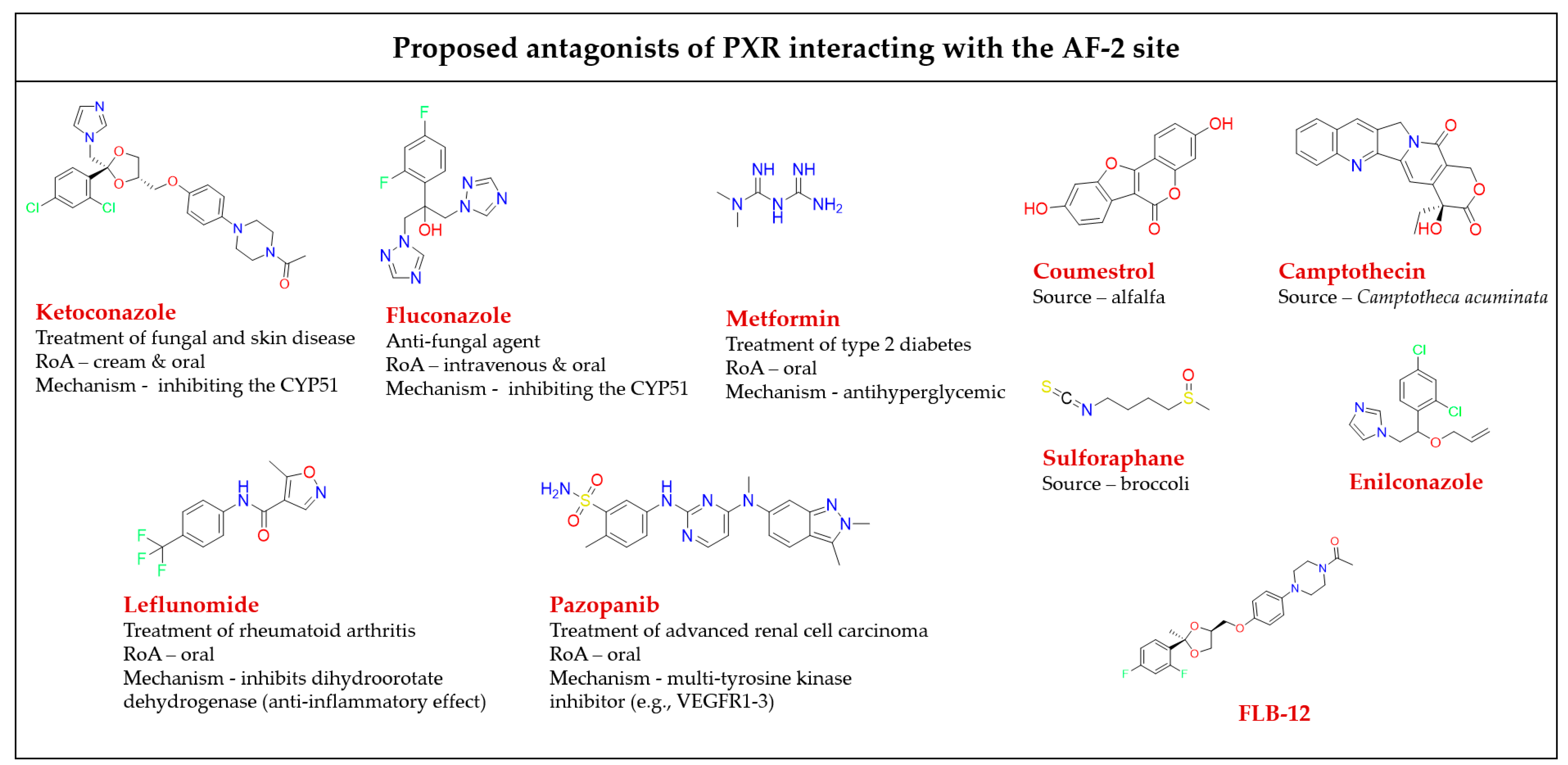
| Molecules | PXR Biological Properties | Binding Site | Reference | |||
|---|---|---|---|---|---|---|
| Efficacy (IC | 50 | ) | Affinity (Ki) | NR Selectivity | ||
| Ketoconazole | 74.4 µM | 55.3 µM | Non-selective | AF-2 | [80] | |
| Coumestrol | 12 µM | 13 µM | Non-selective | AF-2/LBP | [82] | |
| Enilconazole | ~20 µM | NA | NA | AF-2 | [80,83] | [80][83] |
| Fluconazole | ~20 µM | NA | NA | AF-2 | [80,83] | [80][83] |
| FLB-12 | ≥23 µM | NA | Selective | AF-2 | [84] | |
| Leflunomide | 6.8 µM | NA | Non-selective | AF-2 | [83,90] | [83][90] |
| Sulforaphane | 12 µM | 16 µM | Selective | AF-2 | [83,89] | [83][89] |
| Metformin | NA | >1 mM | Non-selective | AF-2 | [87] | |
| Camptothecin | 580 nM | NA | Non-selective | AF-2 | [88] | |
| Pazopanib | 4.1 µM | NA | Selective | AF-2 | [86] | |
| Pimecrolimus | 1.2 µM | NA | Selective | AF-2/LBP | [86] | |
| 73 | 8.3 µM | NA | Selective | AF-2 | [42] | |
References
- Moore, D.D.; Kato, S.; Xie, W.; Mangelsdorf, D.J.; Schmidt, D.R.; Xiao, R.; Kliewer, S.A. International Union of Pharmacology. LXII. The NR1H and NR1I Receptors: Constitutive Androstane Receptor, Pregnene X Receptor, Farnesoid X Receptor α, Farnesoid X Receptor β, Liver X Receptor α, Liver X Receptor β, and Vitamin D Receptor. Pharmacol. Rev. 2006, 58, 742–759.
- Smutny, T.; Mani, S.; Pavek, P. Post-translational and Post-transcriptional Modifications of Pregnane X Receptor (PXR) in Regulation of the Cytochrome P450 Superfamily. Curr. Drug Metab. 2013, 14, 1059–1069.
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589.
- Perrakis, A.; Sixma, T.K. AI revolutions in biology. EMBO Rep. 2021, 22, e54046.
- Monzon, V.; Haft, D.H.; Bateman, A. Folding the unfoldable: Using AlphaFold to explore spurious proteins. Bioinform. Adv. 2022, 2, vbab043.
- Hall, A.; Chanteux, H.; Menochet, K.; Ledecq, M.; Schulze, M.E.D. Designing Out PXR Activity on Drug Discovery Projects: A Review of Structure-Based Methods, Empirical and Computational Approaches. J. Med. Chem. 2021, 64, 6413–6522.
- Wallace, B.D.; Betts, L.; Talmage, G.; Pollet, R.M.; Holman, N.S.; Redinbo, M.R. Structural and Functional Analysis of the Human Nuclear Xenobiotic Receptor PXR in Complex with RXRα. J. Mol. Biol. 2013, 425, 2561–2577.
- Rigalli, J.P.; Theile, D.; Nilles, J.; Weiss, J. Regulation of PXR Function by Coactivator and Corepressor Proteins: Ligand Binding Is Just the Beginning. Cells 2021, 10, 3137.
- Bauer, B.; Hartz, A.M.S.; Fricker, G.; Miller, D.S. Pregnane X Receptor Up-Regulation of P-Glycoprotein Expression and Transport Function at the Blood-Brain Barrier. Mol. Pharmacol. 2004, 66, 413–419.
- Xing, Y.; Yan, J.; Niu, Y. PXR: A center of transcriptional regulation in cancer. Acta Pharm. Sin. B 2020, 10, 197–206.
- Lehmann, J.M.; McKee, D.D.; Watson, M.A.; Willson, T.M.; Moore, J.T.; Kliewer, S.A. The human orphan nuclear receptor PXR is activated by compounds that regulate CYP3A4 gene expression and cause drug interactions. J. Clin. Investig. 1998, 102, 1016–1023.
- Kliewer, S.A.; Moore, J.T.; Wade, L.; Staudinger, J.L.; Watson, M.A.; Jones, S.A.; McKee, D.D.; Oliver, B.B.; Willson, T.M.; Zetterström, R.H.; et al. An Orphan Nuclear Receptor Activated by Pregnanes Defines a Novel Steroid Signaling Pathway. Cell 1998, 92, 73–82.
- Fukuen, S.; Fukuda, T.; Matsuda, H.; Sumida, A.; Yamamoto, I.; Inaba, T.; Azuma, J. Identification of the novel splicing variants for the hPXR in human livers. Biochem. Biophys. Res. Commun. 2002, 298, 433–438.
- Chan, G.N.Y.; Hoque, M.T.; Cummins, C.L.; Bendayan, R. Regulation of P-glycoprotein by orphan nuclear receptors in human brain microvessel endothelial cells. J. Neurochem. 2011, 118, 163–175.
- Atlas, P. Nuclear Receptor Subfamily 1 Group I Member 2. Available online: https://www.proteinatlas.org/ENSG00000144852-NR1I2/tissue (accessed on 22 August 2022).
- Nishimura, M.; Naito, S.; Yokoi, T. Tissue-specific mRNA Expression Profiles of Human Nuclear Receptor Subfamilies. Drug Metab. Pharmacokinet. 2004, 19, 135–149.
- Oladimeji, P.O.; Chen, T. PXR: More Than Just a Master Xenobiotic Receptor. Mol. Pharmacol. 2018, 93, 119–127.
- Creusot, N.; Gassiot, M.; Alaterre, E.; Chiavarina, B.; Grimaldi, M.; Boulahtouf, A.; Toporova, L.; Gerbal-Chaloin, S.; Daujat-Chavanieu, M.; Matheux, A.; et al. The Anti-Cancer Drug Dabrafenib Is a Potent Activator of the Human Pregnane X Receptor. Cells 2020, 9, 1641.
- Feng, F.; Jiang, Q.; Cao, S.; Cao, Y.; Li, R.; Shen, L.; Zhu, H.; Wang, T.; Sun, L.; Liang, E.; et al. Pregnane X receptor mediates sorafenib resistance in advanced hepatocellular carcinoma. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2018, 1862, 1017–1030.
- Gwag, T.; Meng, Z.; Sui, Y.; Helsley, R.N.; Park, S.H.; Wang, S.; Greenberg, R.N.; Zhou, C. Non-nucleoside reverse transcriptase inhibitor efavirenz activates PXR to induce hypercholesterolemia and hepatic steatosis. J. Hepatol. 2019, 70, 930–940.
- Sinz, M.W. Evaluation of pregnane X receptor (PXR)-mediated CYP3A4 drug-drug interactions in drug development. Drug Metab. Rev. 2013, 45, 3–14.
- Shukla, S.J.; Sakamuru, S.; Huang, R.; Moeller, T.A.; Shinn, P.; VanLeer, D.; Auld, D.S.; Austin, C.P.; Xia, M. Identification of Clinically Used Drugs That Activate Pregnane X Receptors. Drug Metab. Dispos. 2011, 39, 151–159.
- Wang, Y.-M.; Ong, S.S.; Chai, S.C.; Chen, T. Role of CAR and PXR in xenobiotic sensing and metabolism. Expert Opin. Drug Metab. Toxicol. 2012, 8, 803–817.
- Ma, X.; Shah, Y.M.; Guo, G.L.; Wang, T.; Krausz, K.W.; Idle, J.R.; Gonzalez, F.J. Rifaximin Is a Gut-Specific Human Pregnane X Receptor Activator. J. Pharmacol. Exp. Ther. 2007, 322, 391–398.
- Daujat-Chavanieu, M.; Gerbal-Chaloin, S. Regulation of CAR and PXR Expression in Health and Disease. Cells 2020, 9, 2395.
- Pondugula, S.R.; Pavek, P.; Mani, S. Pregnane X Receptor and Cancer: Context-Specificity is Key. Nucl. Recep. Res. 2016, 3, 101198.
- Bansard, L.; Bouvet, O.; Moutin, E.; Le Gall, G.; Giammona, A.; Pothin, E.; Bacou, M.; Hassen-Khodja, C.; Bordignon, B.; Bourgaux, J.F.; et al. Niclosamide induces miR-148a to inhibit PXR and sensitize colon cancer stem cells to chemotherapy. Stem Cell Rep. 2022, 17, 835–848.
- Wang, H.; Venkatesh, M.; Li, H.; Goetz, R.; Mukherjee, S.; Biswas, A.; Zhu, L.; Kaubisch, A.; Wang, L.; Pullman, J.; et al. Pregnane X receptor activation induces FGF19-dependent tumor aggressiveness in humans and mice. J. Clin. Investig. 2011, 121, 3220–3232.
- Raynal, C.; Pascussi, J.-M.; Leguelinel, G.; Breuker, C.; Kantar, J.; Lallemant, B.; Poujol, S.; Bonnans, C.; Joubert, D.; Hollande, F.; et al. Pregnane × Receptor (PXR) expression in colorectal cancer cells restricts irinotecan chemosensitivity through enhanced SN-38 glucuronidation. Mol. Cancer 2010, 9, 46.
- Planque, C.; Rajabi, F.; Grillet, F.; Finetti, P.; Bertucci, F.; Gironella, M.; Lozano, J.J.; Beucher, B.; Giraud, J.; Garambois, V.; et al. Pregnane X-receptor promotes stem cell-mediated colon cancer relapse. Oncotarget 2016, 7, 56558–56573.
- Niu, X.; Wu, T.; Li, G.; Gu, X.; Tian, Y.; Cui, H. Insights into the critical role of the PXR in preventing carcinogenesis and chemotherapeutic drug resistance. Int. J. Biol. Sci. 2022, 18, 742–759.
- Lynch, C.; Sakamuru, S.; Huang, R.; Niebler, J.; Ferguson, S.S.; Xia, M. Characterization of human pregnane X receptor activators identified from a screening of the Tox21 compound library. Biochem. Pharmacol. 2021, 184, 114368.
- Zhang, J.; Pavek, P.; Kamaraj, R.; Ren, L.; Zhang, T. Dietary phytochemicals as modulators of human pregnane X receptor. Crit. Rev. Food Sci. Nutr. 2021, 1–23.
- Karpale, M.; Hukkanen, J.; Hakkola, J. Nuclear Receptor PXR in Drug-Induced Hypercholesterolemia. Cells 2022, 11, 313.
- Sayaf, K.; Zanotto, I.; Russo, F.P.; Gabbia, D.; De Martin, S. The Nuclear Receptor PXR in Chronic Liver Disease. Cells 2021, 11, 61.
- Pondugula, S.R.; Mani, S. Pregnane xenobiotic receptor in cancer pathogenesis and therapeutic response. Cancer Lett. 2013, 328, 1–9.
- Chai, S.C.; Wright, W.C.; Chen, T. Strategies for developing pregnane X receptor antagonists: Implications from metabolism to cancer. Med. Res. Rev. 2020, 40, 1061–1083.
- Biswas, A.; Mani, S.; Redinbo, M.R.; Krasowski, M.D.; Li, H.; Ekins, S. Elucidating the ‘Jekyll and Hyde’ Nature of PXR: The Case for Discovering Antagonists or Allosteric Antagonists. Pharm. Res. 2009, 26, 1807–1815.
- Li, Y.; Lin, W.; Wright, W.C.; Chai, S.C.; Wu, J.; Chen, T. Building a Chemical Toolbox for Human Pregnane X Receptor Research: Discovery of Agonists, Inverse Agonists, and Antagonists Among Analogs Based on the Unique Chemical Scaffold of SPA70. J. Med. Chem. 2021, 64, 1733–1761.
- Watkins, R.E.; Wisely, G.B.; Moore, L.B.; Collins, J.L.; Lambert, M.H.; Williams, S.P.; Willson, T.M.; Kliewer, S.A.; Redinbo, M.R. The human nuclear xenobiotic receptor PXR: Structural determinants of directed promiscuity. Science 2001, 292, 2329–2333.
- Lin, W.; Wang, Y.M.; Chai, S.C.; Lv, L.; Zheng, J.; Wu, J.; Zhang, Q.; Wang, Y.D.; Griffin, P.R.; Chen, T. SPA70 is a potent antagonist of human pregnane X receptor. Nat. Commun. 2017, 8, 741.
- Mustonen, E.-K.; Pantsar, T.; Rashidian, A.; Reiner, J.; Schwab, M.; Laufer, S.; Burk, O. Target Hopping from Protein Kinases to PXR: Identification of Small-Molecule Protein Kinase Inhibitors as Selective Modulators of Pregnane X Receptor from TüKIC Library. Cells 2022, 11, 1299.
- Ong, S.S. Pregnane X Receptor in Drug Development; IntechOpen: London, UK, 2011.
- Motta, S.; Callea, L.; Giani Tagliabue, S.; Bonati, L. Exploring the PXR ligand binding mechanism with advanced Molecular Dynamics methods. Sci. Rep. 2018, 8, 16207.
- Huang, P.; Chandra, V.; Rastinejad, F. Structural Overview of the Nuclear Receptor Superfamily: Insights into Physiology and Therapeutics. Annu. Rev. Physiol. 2010, 72, 247–272.
- Wu, B.; Li, S.; Dong, D. 3D structures and ligand specificities of nuclear xenobiotic receptors CAR, PXR and VDR. Drug Discov. Today 2013, 18, 574–581.
- Liu, T.; Beck, J.P.; Hao, J. A concise review on hPXR ligand-recognizing residues and structure-based strategies to alleviate hPXR transactivation risk. RSC Med. Chem. 2022, 13, 129–137.
- Huber, A.D.; Wright, W.C.; Lin, W.; Majumder, K.; Low, J.A.; Wu, J.; Buchman, C.D.; Pintel, D.J.; Chen, T. Mutation of a single amino acid of pregnane X receptor switches an antagonist to agonist by altering AF-2 helix positioning. Cell Mol. Life Sci. 2021, 78, 317–335.
- Huber, A.D.; Li, Y.; Lin, W.; Galbraith, A.N.; Mishra, A.; Porter, S.N.; Wu, J.; Florke Gee, R.R.; Zhuang, W.; Pruett-Miller, S.M.; et al. SJPYT-195: A Designed Nuclear Receptor Degrader That Functions as a Molecular Glue Degrader of GSPT1. ACS Med. Chem. Lett. 2022, 13, 1311–1320.
- Zorba, A.; Nguyen, C.; Xu, Y.; Starr, J.; Borzilleri, K.; Smith, J.; Zhu, H.; Farley, K.A.; Ding, W.; Schiemer, J.; et al. Delineating the role of cooperativity in the design of potent PROTACs for BTK. Proc. Natl. Acad. Sci. USA 2018, 115, E7285–E7292.
- Shimokawa, K.; Shibata, N.; Sameshima, T.; Miyamoto, N.; Ujikawa, O.; Nara, H.; Ohoka, N.; Hattori, T.; Cho, N.; Naito, M. Targeting the Allosteric Site of Oncoprotein BCR-ABL as an Alternative Strategy for Effective Target Protein Degradation. ACS Med. Chem. Lett. 2017, 8, 1042–1047.
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Meng, E.C.; Couch, G.S.; Croll, T.I.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci. 2021, 30, 70–82.
- De Smet, F.; Christopoulos, A.; Carmeliet, P. Allosteric targeting of receptor tyrosine kinases. Nat. Biotechnol. 2014, 32, 1113–1120.
- Gregory, K.J.; Sexton, P.M.; Christopoulos, A. Overview of receptor allosterism. Curr. Protoc. Pharmacol. 2000, 11, 1–21.
- Christopoulos, A.; Kenakin, T. G protein-coupled receptor allosterism and complexing. Pharmacol. Rev. 2002, 54, 323–374.
- Smith, N.J.; Milligan, G. Allostery at G Protein-Coupled Receptor Homo- and Heteromers: Uncharted Pharmacological Landscapes. Pharmacol. Rev. 2010, 62, 701–725.
- Conn, P.J.; Christopoulos, A.; Lindsley, C.W. Allosteric modulators of GPCRs: A novel approach for the treatment of CNS disorders. Nat. Rev. Drug Discov. 2009, 8, 41–54.
- Christopoulos, A. Advances in G Protein-Coupled Receptor Allostery: From Function to Structure. Mol. Pharmacol. 2014, 86, 463–478.
- Lewis, J.A.; Scott, S.A.; Lavieri, R.; Buck, J.R.; Selvy, P.E.; Stoops, S.L.; Armstrong, M.D.; Brown, H.A.; Lindsley, C.W. Design and synthesis of isoform-selective phospholipase D (PLD) inhibitors. Part I: Impact of alternative halogenated privileged structures for PLD1 specificity. Bioorg. Med. Chem. Lett. 2009, 19, 1916–1920.
- Cao, A.M.; Quast, R.B.; Fatemi, F.; Rondard, P.; Pin, J.P.; Margeat, E. Allosteric modulators enhance agonist efficacy by increasing the residence time of a GPCR in the active state. Nat. Commun. 2021, 12, 5426.
- Reddy, G.S.; Kamaraj, R.; Hossain, K.A.; Kumar, J.S.; Thirupataiah, B.; Medishetti, R.; Sushma Sri, N.; Misra, P.; Pal, M. Amberlyst-15 catalysed synthesis of novel indole derivatives under ultrasound irradiation: Their evaluation as serotonin 5-HT2C receptor agonists. Bioorg. Chem. 2021, 116, 105380.
- Burford, N.T.; Clark, M.J.; Wehrman, T.S.; Gerritz, S.W.; Banks, M.; O’Connell, J.; Traynor, J.R.; Alt, A. Discovery of positive allosteric modulators and silent allosteric modulators of the mu-opioid receptor. Proc. Natl. Acad. Sci. USA 2013, 110, 10830–10835.
- Kenakin, T.; Strachan, R.T. PAM-Antagonists: A Better Way to Block Pathological Receptor Signaling? Trends Pharmacol. Sci. 2018, 39, 748–765.
- Fasciani, I.; Petragnano, F.; Aloisi, G.; Marampon, F.; Carli, M.; Scarselli, M.; Maggio, R.; Rossi, M. Allosteric Modulators of G Protein-Coupled Dopamine and Serotonin Receptors: A New Class of Atypical Antipsychotics. Pharmaceuticals 2020, 13, 388.
- Allosteric Database (ASD). Nuclear Hormone Receptor >> 202 Modulators. Available online: http://mdl.shsmu.edu.cn/ASD/module/modulators/modulators.jsp (accessed on 14 May 2022).
- Changeux, J.P.; Christopoulos, A. Allosteric Modulation as a Unifying Mechanism for Receptor Function and Regulation. Cell 2016, 166, 1084–1102.
- Moore, T.W.; Mayne, C.G.; Katzenellenbogen, J.A. Minireview: Not Picking Pockets: Nuclear Receptor Alternate-Site Modulators (NRAMs). Mol. Endocrinol. 2010, 24, 683–695.
- Chen, Y.; Li, J.; Wu, Z.; Liu, G.; Li, H.; Tang, Y.; Li, W. Computational Insight into the Allosteric Activation Mechanism of Farnesoid X Receptor. J. Chem. Inf. Model. 2020, 60, 1540–1550.
- Gabler, M.; Kramer, J.; Schmidt, J.; Pollinger, J.; Weber, J.; Kaiser, A.; Lohr, F.; Proschak, E.; Schubert-Zsilavecz, M.; Merk, D. Allosteric modulation of the farnesoid X receptor by a small molecule. Sci. Rep. 2018, 8, 6846.
- Delfosse, V.; Dendele, B.; Huet, T.; Grimaldi, M.; Boulahtouf, A.; Gerbal-Chaloin, S.; Beucher, B.; Roecklin, D.; Muller, C.; Rahmani, R.; et al. Synergistic activation of human pregnane X receptor by binary cocktails of pharmaceutical and environmental compounds. Nat. Commun. 2015, 6, 8089.
- Wang, Y.; Chirgadze, N.Y.; Briggs, S.L.; Khan, S.; Jensen, E.V.; Burris, T.P. A second binding site for hydroxytamoxifen within the coactivator-binding groove of estrogen receptor β. Proc. Natl. Acad. Sci. USA 2006, 103, 9908–9911.
- Hughes, T.S.; Giri, P.K.; de Vera, I.M.; Marciano, D.P.; Kuruvilla, D.S.; Shin, Y.; Blayo, A.L.; Kamenecka, T.M.; Burris, T.P.; Griffin, P.R.; et al. An alternate binding site for PPARgamma ligands. Nat. Commun. 2014, 5, 3571.
- Delfosse, V.; Huet, T.; Harrus, D.; Granell, M.; Bourguet, M.; Gardia-Parège, C.; Chiavarina, B.; Grimaldi, M.; Le Mével, S.; Blanc, P.; et al. Mechanistic insights into the synergistic activation of the RXR–PXR heterodimer by endocrine disruptor mixtures. Proc. Natl. Acad. Sci. USA 2021, 118, e2020551118.
- Lane, J.R.; Sexton, P.M.; Christopoulos, A. Bridging the gap: Bitopic ligands of G-protein-coupled receptors. Trends Pharmacol. Sci. 2013, 34, 59–66.
- Zhang, C.; Wu, J.; Chen, Q.; Tan, H.; Huang, F.; Guo, J.; Zhang, X.; Yu, H.; Shi, W. Allosteric binding on nuclear receptors: Insights on screening of non-competitive endocrine-disrupting chemicals. Environ. Int. 2022, 159, 107009.
- Pavek, P. Pregnane X Receptor (PXR)-Mediated Gene Repression and Cross-Talk of PXR with Other Nuclear Receptors via Coactivator Interactions. Front. Pharmacol. 2016, 7, 456.
- La Sala, G.; Gunnarsson, A.; Edman, K.; Tyrchan, C.; Hogner, A.; Frolov, A.I. Unraveling the Allosteric Cross-Talk between the Coactivator Peptide and the Ligand-Binding Site in the Glucocorticoid Receptor. J. Chem. Inf. Model. 2021, 61, 3667–3680.
- Fischer, A.; Smieško, M. Allosteric Binding Sites On Nuclear Receptors: Focus On Drug Efficacy and Selectivity. Int. J. Mol. Sci. 2020, 21, 534.
- Wärnmark, A.; Treuter, E.; Wright, A.P.H.; Gustafsson, J.-A.k. Activation Functions 1 and 2 of Nuclear Receptors: Molecular Strategies for Transcriptional Activation. Mol. Endocrinol. 2003, 17, 1901–1909.
- Wang, H.; Huang, H.; Li, H.; Teotico, D.G.; Sinz, M.; Baker, S.D.; Staudinger, J.; Kalpana, G.; Redinbo, M.R.; Mani, S. Activated pregnenolone X-receptor is a target for ketoconazole and its analogs. Clin. Cancer Res. 2007, 13, 2488–2495.
- Huang, H.; Wang, H.; Sinz, M.; Zoeckler, M.; Staudinger, J.; Redinbo, M.R.; Teotico, D.G.; Locker, J.; Kalpana, G.V.; Mani, S. Inhibition of drug metabolism by blocking the activation of nuclear receptors by ketoconazole. Oncogene 2007, 26, 258–268.
- Wang, H.; Li, H.; Moore, L.B.; Johnson, M.D.; Maglich, J.M.; Goodwin, B.; Ittoop, O.R.; Wisely, B.; Creech, K.; Parks, D.J.; et al. The phytoestrogen coumestrol is a naturally occurring antagonist of the human pregnane X receptor. Mol. Endocrinol. 2008, 22, 838–857.
- Ekins, S.; Kholodovych, V.; Ai, N.; Sinz, M.; Gal, J.; Gera, L.; Welsh, W.J.; Bachmann, K.; Mani, S. Computational discovery of novel low micromolar human pregnane X receptor antagonists. Mol. Pharmacol. 2008, 74, 662–672.
- Venkatesh, M.; Wang, H.; Cayer, J.; Leroux, M.; Salvail, D.; Das, B.; Wrobel, J.E.; Mani, S. In vivo and in vitro characterization of a first-in-class novel azole analog that targets pregnane X receptor activation. Mol. Pharmacol. 2011, 80, 124–135.
- Ekins, S.; Chang, C.; Mani, S.; Krasowski, M.D.; Reschly, E.J.; Iyer, M.; Kholodovych, V.; Ai, N.; Welsh, W.J.; Sinz, M.; et al. Human pregnane X receptor antagonists and agonists define molecular requirements for different binding sites. Mol. Pharmacol. 2007, 72, 592–603.
- Burk, O.; Kuzikov, M.; Kronenberger, T.; Jeske, J.; Keminer, O.; Thasler, W.E.; Schwab, M.; Wrenger, C.; Windshügel, B. Identification of approved drugs as potent inhibitors of pregnane X receptor activation with differential receptor interaction profiles. Arch. Toxicol. 2018, 92, 1435–1451.
- Krausova, L.; Stejskalova, L.; Wang, H.; Vrzal, R.; Dvorak, Z.; Mani, S.; Pavek, P. Metformin suppresses pregnane X receptor (PXR)-regulated transactivation of CYP3A4 gene. Biochem. Pharmacol. 2011, 82, 1771–1780.
- Chen, Y.; Tang, Y.; Robbins, G.T.; Nie, D. Camptothecin attenuates cytochrome P450 3A4 induction by blocking the activation of human pregnane X receptor. J. Pharmacol. Exp. Ther. 2010, 334, 999–1008.
- Zhou, C.; Poulton, E.J.; Grun, F.; Bammler, T.K.; Blumberg, B.; Thummel, K.E.; Eaton, D.L. The dietary isothiocyanate sulforaphane is an antagonist of the human steroid and xenobiotic nuclear receptor. Mol. Pharmacol. 2007, 71, 220–229.
- Carazo, A.; Dusek, J.; Holas, O.; Skoda, J.; Hyrsova, L.; Smutny, T.; Soukup, T.; Dosedel, M.; Pávek, P. Teriflunomide Is an Indirect Human Constitutive Androstane Receptor (CAR) Activator Interacting with Epidermal Growth Factor (EGF) Signaling. Front. Pharmacol. 2018, 9, 993.
- Kronenberger, T.; Keminer, O.; Wrenger, C.; Windshügel, B. Nuclear Receptor Modulators—Current Approaches and Future Perspectives. In Drug Discovery and Development: From Molecules to Medicine; InTech: London, UK, 2015.