 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Chi Teng Vong | -- | 2953 | 2022-10-12 11:48:25 | | | |
| 2 | Conner Chen | -5 word(s) | 2948 | 2022-10-12 11:56:45 | | | | |
| 3 | Conner Chen | + 7 word(s) | 2955 | 2022-10-12 11:59:17 | | |
Video Upload Options
Type 2 diabetes mellitus (T2DM) is a chronic metabolic disease, which is characterized by hyperglycemia, chronic insulin resistance, progressive decline in β-cell function, and defect in insulin secretion. It has become one of the leading causes of death worldwide. There is no cure for T2DM, but it can be treated, and blood glucose levels can be controlled. It has been reported that diabetic patients may suffer from the adverse effects of conventional medicine. Therefore, alternative therapy, such as traditional Chinese medicine (TCM), can be used to manage and treat diabetes. Glycyrrhizic acid (GL) and its derivatives are suggested to be promising candidates for the treatment of T2DM and its complications. It is the principal bioactive constituent in licorice, one type of TCM.
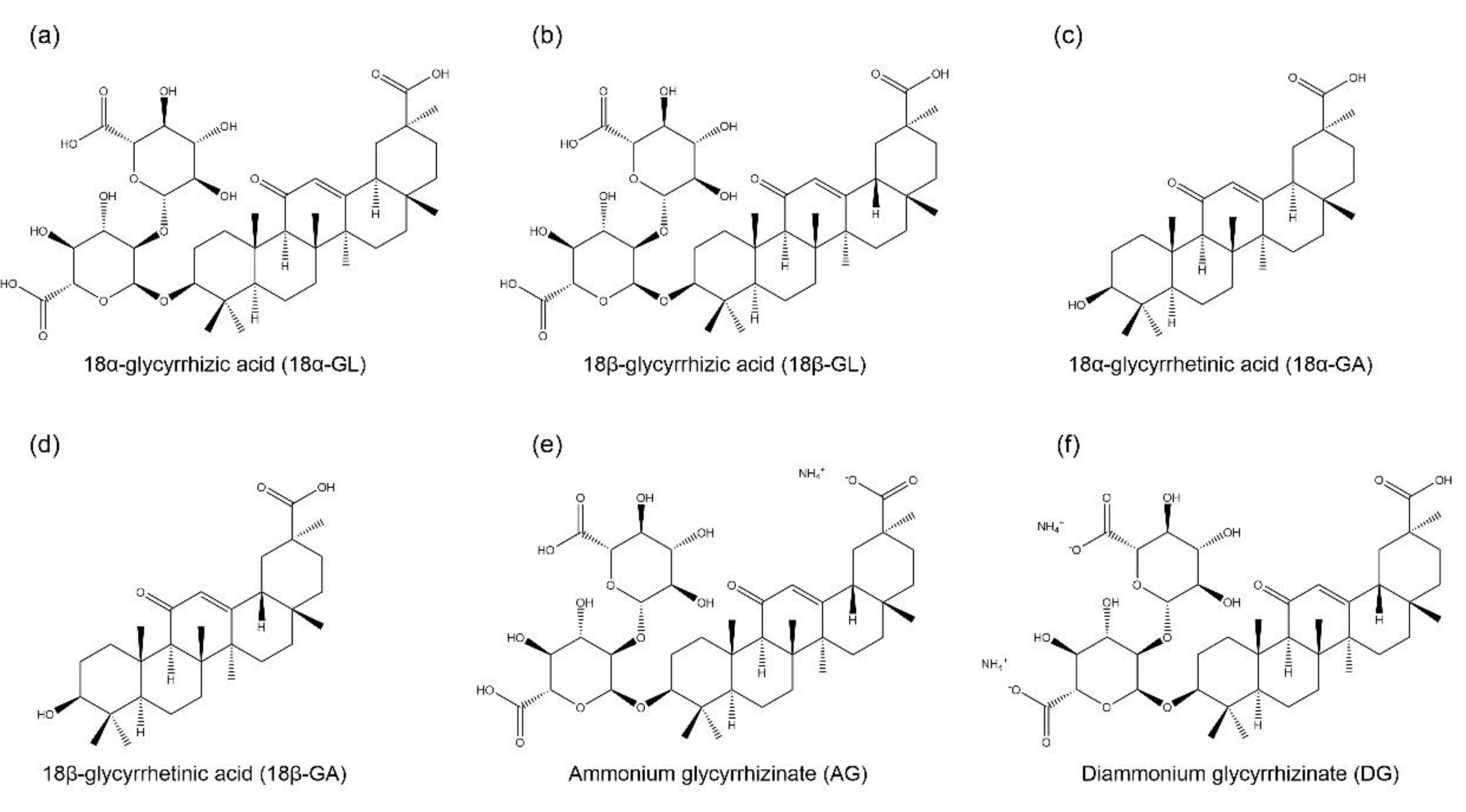
1. GL and Its Derivatives
2. Anti-Diabetic Mechanisms of GL and Its Derivatives in T2DM
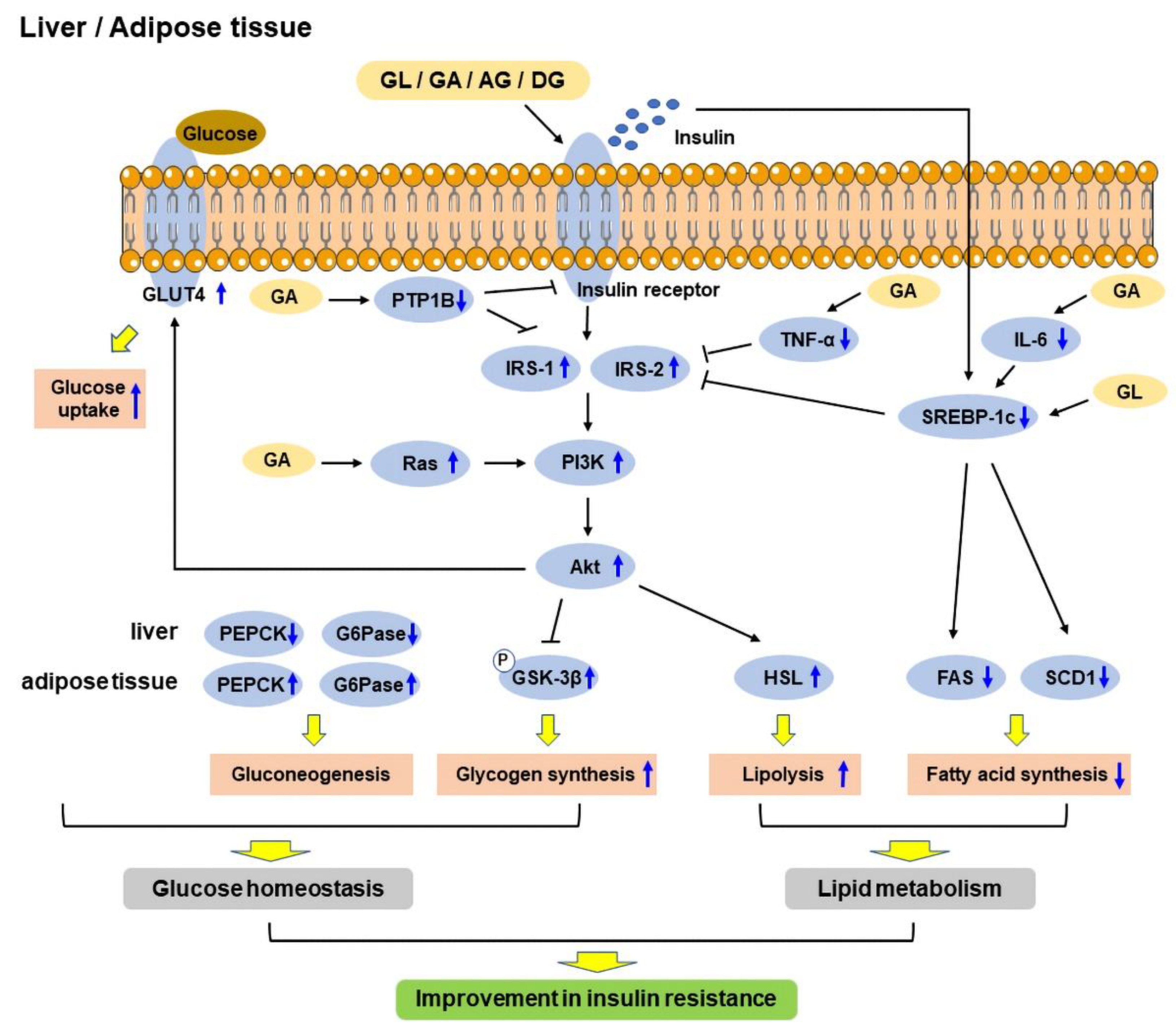
2.1. Insulin Resistance
2.2. Glucose Tolerance and Homeostasis
2.3. Lipid Metabolism
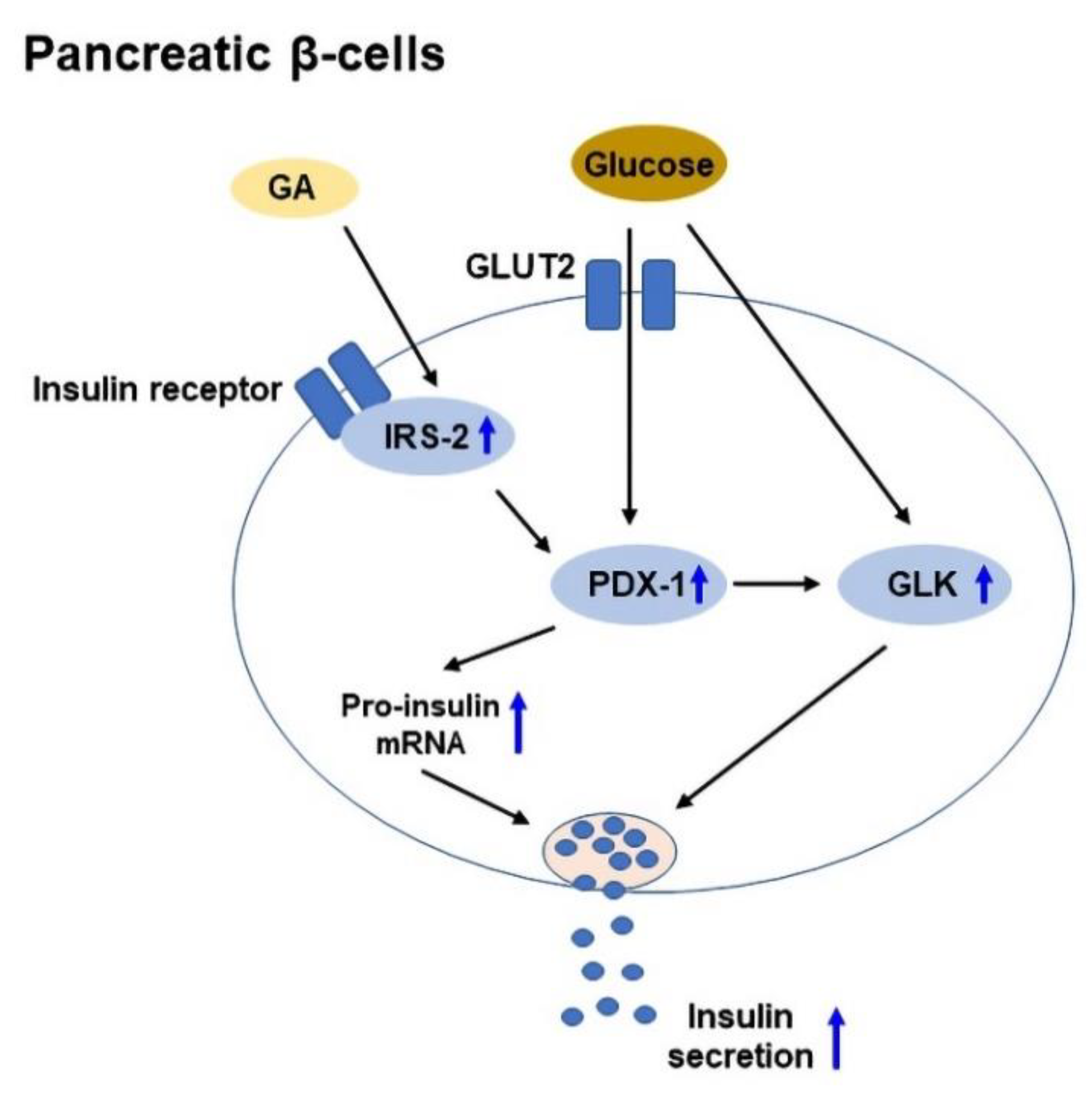
2.4. Insulin Secretion and Protection of Pancreatic β-Cells
References
- Pastorino, G.; Cornara, L.; Soares, S.; Rodrigues, F.; Oliveira, M.B.P.P. Liquorice (Glycyrrhiza glabra): A phytochemical and pharmacological review. Phytother. Res. 2018, 32, 2323–2339.
- Newell-Price, J.D.C. Cushing Disease. In The Pituitary, 4th ed.; Melmed, S., Ed.; Academic Press: Cambridge, MA, USA, 2017; pp. 515–571.
- Sabbioni, C.; Mandrioli, R.; Ferranti, A.; Bugamelli, F.; Saracino, M.A.; Forti, G.C.; Fanali, S.; Raggi, M.A. Separation and analysis of glycyrrhizin, 18beta-glycyrrhetic acid and 18alpha-glycyrrhetic acid in liquorice roots by means of capillary zone electrophoresis. J. Chromatogr. A 2005, 1081, 65–71.
- Graebin, C.S. The Pharmacological Activities of Glycyrrhizinic Acid (“Glycyrrhizin”) and Glycyrrhetinic Acid. In Sweeteners: Pharmacology, Biotechnology, and Applications, 1st ed.; Merillon, J.-M., Ramawat, K.G., Eds.; Springer International Publishing: New York, NY, USA, 2017; pp. 245–261.
- Yang, L.; Jiang, Y.; Zhang, Z.; Hou, J.; Tian, S.; Liu, Y. The anti-diabetic activity of licorice, a widely used Chinese herb. J. Ethnopharmacol. 2020, 263, 113216.
- Takeda, S.; Ishthara, K.; Wakui, Y.; Amagaya, S.; Maruno, M.; Akao, T.; Kobashi, K. Bioavailability study of glycyrrhetic acid after oral administration of glycyrrhizin in rats; relevance to the intestinal bacterial hydrolysis. J. Pharm. Pharmacol. 1996, 48, 902–905.
- Akao, T.; Hayashi, T.; Kobashi, K.; Kanaoka, M.; Kato, H.; Kobayashi, M.; Takeda, S.; Oyama, T. Intestinal bacterial hydrolysis is indispensable to absorption of 18 beta-glycyrrhetic acid after oral administration of glycyrrhizin in rats. J. Pharm. Pharmacol. 1994, 46, 135–137.
- Yang, J.; Zhou, L.; Wang, J.; Wang, G.; Davey, A.K. The disposition of diammonium glycyrrhizinate and glycyrrhetinic acid in the isolated perfused rat intestine and liver. Planta Med. 2008, 74, 1351–1356.
- Jin, S.; Fu, S.; Han, J.; Jin, S.; Lv, Q.; Lu, Y.; Qi, J.; Wu, W.; Yuan, H. Improvement of oral bioavailability of glycyrrhizin by sodium deoxycholate/phospholipid-mixed nanomicelles. J. Drug Target. 2012, 20, 615–622.
- Fry, J.C. Natural low-calorie sweeteners. In Natural Food Additives, Ingredients and Flavourings, 1st ed.; Baines, D., Seal, R., Eds.; Woodhead Publishing: Sawston, UK, 2012; pp. 41–75.
- Galicia-Garcia, U.; Benito-Vicente, A.; Jebari, S.; Larrea-Sebal, A.; Siddiqi, H.; Uribe, K.B.; Ostolaza, H.; Martin, C. Pathophysiology of type 2 diabetes mellitus. Int. J. Mol. Sci. 2020, 21, 6275.
- Sun, X.; Duan, X.; Wang, C.; Liu, Z.; Sun, P.; Huo, X.; Ma, X.; Sun, H.; Liu, K.; Meng, Q. Protective effects of glycyrrhizic acid against non-alcoholic fatty liver disease in mice. Eur. J. Pharmacol. 2017, 806, 75–82.
- Sil, R.; Ray, D.; Chakraborti, A.S. Glycyrrhizin ameliorates insulin resistance, hyperglycemia, dyslipidemia and oxidative stress in fructose-induced metabolic syndrome-X in rat model. Indian J. Exp. Biol. 2013, 51, 129–138.
- Cheng, H.S.; Yaw, H.P.; Ton, S.H.; Choy, S.M.; Kong, J.M.; Abdul Kadir, K. Glycyrrhizic acid prevents high calorie diet-induced metabolic aberrations despite the suppression of peroxisome proliferator-activated receptor gamma expression. Nutrition 2016, 32, 995–1001.
- Ko, B.S.; Jang, J.S.; Hong, S.M.; Sung, S.R.; Lee, J.E.; Lee, M.Y.; Jeon, W.K.; Park, S. Changes in components, glycyrrhizin and glycyrrhetinic acid, in raw Glycyrrhiza uralensis Fisch, modify insulin sensitizing and insulinotropic actions. Biosci. Biotechnol. Biochem. 2007, 71, 1452–1461.
- Boucher, J.; Kleinridders, A.; Kahn, C.R. Insulin receptor signaling in normal and insulin-resistant states. Cold Spring Harb. Perspect. Biol. 2014, 6, a009191.
- National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK). Insulin Resistance & Prediabetes. Available online: https://www.niddk.nih.gov/health-information/diabetes/overview/what-is-diabetes/prediabetes-insulin-resistance (accessed on 12 June 2022).
- Fonseca, V.A. Defining and characterizing the progression of type 2 diabetes. Diabetes Care 2009, 32 (Suppl. 2), S151–S156.
- Abo El-Magd, N.F.; El-Mesery, M.; El-Karef, A.; El-Shishtawy, M.M. Glycyrrhizin ameliorates high fat diet-induced obesity in rats by activating NrF2 pathway. Life Sci. 2018, 193, 159–170.
- Liu, L.; Jiang, Y.; Steinle, J.J. Inhibition of HMGB1 protects the retina from ischemia-reperfusion, as well as reduces insulin resistance proteins. PLoS ONE 2017, 12, e0178236.
- Chia, Y.Y.; Ton, S.H.; Kadir, A.K. Effects of glycyrrhizic acid on peroxisome proliferator-activated receptor gamma (PPARgamma), lipoprotein lipase (LPL), serum lipid and HOMA-IR in rats. PPAR Res. 2010, 2010, 530265.
- Cheng, H.S.; Kong, J.M.; Ng, A.X.; Chan, W.K.; Ton, S.H.; Abdul Kadir, K. Novel inhibitory effects of glycyrrhizic acid on the accumulation of advanced glycation end product and its receptor expression. Nat. Prod. Bioprospect. 2014, 4, 325–333.
- Li, Y.; Hou, H.; Wang, X.; Dai, X.; Zhang, W.; Tang, Q.; Dong, Y.; Yan, C.; Wang, B.; Li, Z.; et al. Diammonium glycyrrhizinate ameliorates obesity through modulation of gut microbiota-conjugated BAs-FXR signaling. Front. Pharmacol. 2021, 12, 796590.
- Zhang, Y.; Yang, S.; Zhang, M.; Wang, Z.; He, X.; Hou, Y.; Bai, G. Glycyrrhetinic acid improves insulin-response pathway by regulating the balance between the Ras/MAPK and PI3K/Akt pathways. Nutrients 2019, 11, 604.
- Fatimah, M.; Malik, M.; Mushtaq, S.; Sarfraz, J.; Mushtaq, Z.; Chiragh, S. Dose dependent effect of glycyrrhizin on glycaemic control of type 2 diabetic rats. Khyber Med. Univ. J. 2020, 12, 121–125.
- Chia, Y.Y.; Ton, S.H.; Kadir, K.B. Effects of glycyrrhizic acid on 11 beta-hydroxysteroid dehydrogenase (11 betaHSD1 and 2) activities and HOMA-IR in rats at different treatment periods. Exp. Clin. Endocrinol. Diabetes 2010, 118, 617–624.
- Chia, Y.Y.; Liong, S.Y.; Ton, S.H.; Kadir, K.B. Amelioration of glucose homeostasis by glycyrrhizic acid through gluconeogenesis rate-limiting enzymes. Eur. J. Pharmacol. 2012, 677, 197–202.
- Chandramouli, C.; Ting, Y.S.; Lyn, L.Y.; Ha, T.S.; Kadir, K.A. Glycyrrhizic acid improves lipid and glucose metabolism in high-sucrose-fed rats. J. Endocrinol. Metab. 2011, 1, 125–141.
- Takii, H.; Kometani, T.; Nishimura, T.; Nakae, T.; Okada, S.; Fushiki, T. Antidiabetic effect of glycyrrhizin in genetically diabetic KK-Ay mice. Biol. Pharm. Bull. 2001, 24, 484–487.
- Lim, W.Y.; Chia, Y.Y.; Liong, S.Y.; Ton, S.H.; Kadir, K.A.; Husain, S.N. Lipoprotein lipase expression, serum lipid and tissue lipid deposition in orally-administered glycyrrhizic acid-treated rats. Lipids Health Dis. 2009, 8, 31.
- Muniyappa, R.; Madan, R.; Varghese, R.T. Assessing Insulin Sensitivity and Resistance in Humans. In Endotext; Feingold, K.R., Anawalt, B., Boyce, A., Chrousos, G., de Herder, W.W., Dhatariya, K., Dungan, K., Hershman, J.M., Hofland, J., Kalra, S., et al., Eds.; MD Text: South Dartmouth, MA, USA, 2021.
- Matthews, D.R.; Hosker, J.P.; Rudenski, A.S.; Naylor, B.A.; Treacher, D.F.; Turner, R.C. Homeostasis model assessment: Insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia 1985, 28, 412–419.
- Katsuki, A.; Sumida, Y.; Gabazza, E.C.; Murashima, S.; Urakawa, H.; Morioka, K.; Kitagawa, N.; Tanaka, T.; Araki-Sasaki, R.; Hori, Y.; et al. QUICKI is useful for following improvements in insulin sensitivity after therapy in patients with type 2 diabetes mellitus. J. Clin. Endocrinol. Metab. 2002, 87, 2906–2908.
- Sasaki, N.; Ozono, R.; Higashi, Y.; Maeda, R.; Kihara, Y. Association of insulin resistance, plasma glucose level, and serum insulin level with hypertension in a population with different stages of impaired glucose metabolism. J. Am. Heart Assoc. 2020, 9, e015546.
- Chaour, M.; Theroux, P.; Gilfix, B.M.; Campeau, L.; Lesperance, J.; Ghitescu, M.; Gelinas, F.; Solymoss, B.C. ‘True’ fasting serum insulin level, insulin resistance syndrome and coronary artery disease. Coron. Artery Dis. 1997, 8, 683–688.
- Zhang, H.; Zhang, C. Adipose “talks” to distant organs to regulate insulin sensitivity and vascular function. Obesity 2010, 18, 2071–2076.
- Hoffstedt, J.; Arner, E.; Wahrenberg, H.; Andersson, D.P.; Qvisth, V.; Lofgren, P.; Ryden, M.; Thorne, A.; Wiren, M.; Palmer, M.; et al. Regional impact of adipose tissue morphology on the metabolic profile in morbid obesity. Diabetologia 2010, 53, 2496–2503.
- Vidal-Puig, A.J.; Considine, R.V.; Jimenez-Linan, M.; Werman, A.; Pories, W.J.; Caro, J.F.; Flier, J.S. Peroxisome proliferator-activated receptor gene expression in human tissues. Effects of obesity, weight loss, and regulation by insulin and glucocorticoids. J. Clin. Investig. 1997, 99, 2416–2422.
- Leonardini, A.; Laviola, L.; Perrini, S.; Natalicchio, A.; Giorgino, F. Cross-talk between PPARgamma and insulin signaling and modulation of insulin sensitivity. PPAR Res. 2009, 2009, 818945.
- Santoleri, D.; Titchenell, P.M. Resolving the paradox of hepatic insulin resistance. Cell. Mol. Gastroenterol. Hepatol. 2019, 7, 447–456.
- Sesti, G.; Federici, M.; Hribal, M.L.; Lauro, D.; Sbraccia, P.; Lauro, R. Defects of the insulin receptor substrate (IRS) system in human metabolic disorders. FASEB J. 2001, 15, 2099–2111.
- Goldstein, B.J. Protein-tyrosine phosphatase 1B (PTP1B): A novel therapeutic target for type 2 diabetes mellitus, obesity and related states of insulin resistance. Curr. Drug Targets Immune Endocr. Metabol. Disord. 2001, 1, 265–275.
- Combs, A.P. Recent advances in the discovery of competitive protein tyrosine phosphatase 1B inhibitors for the treatment of diabetes, obesity, and cancer. J. Med. Chem. 2010, 53, 2333–2344.
- Seong, S.H.; Nguyen, D.H.; Wagle, A.; Woo, M.H.; Jung, H.A.; Choi, J.S. Experimental and computational study to reveal the potential of non-polar constituents from Hizikia fusiformis as dual protein tyrosine phosphatase 1B and alpha-glucosidase inhibitors. Mar. Drugs 2019, 17, 302.
- Na, M.; Cui, L.; Min, B.S.; Bae, K.; Yoo, J.K.; Kim, B.Y.; Oh, W.K.; Ahn, J.S. Protein tyrosine phosphatase 1B inhibitory activity of triterpenes isolated from Astilbe oreana. Bioorg. Med. Chem. Lett. 2006, 16, 3273–3276.
- De-la-Cruz-Martinez, L.; Duran-Becerra, C.; Gonzalez-Andrade, M.; Paez-Franco, J.C.; German-Acacio, J.M.; Espinosa-Chavez, J.; Torres-Valencia, J.M.; Perez-Villanueva, J.; Palacios-Espinosa, J.F.; Soria-Arteche, O.; et al. Indole- and pyrazole-glycyrrhetinic acid derivatives as PTP1B inhibitors: Synthesis, in vitro and in silico studies. Molecules 2021, 26, 4375.
- Moller, N.; Jorgensen, J.O. Effects of growth hormone on glucose, lipid, and protein metabolism in human subjects. Endocr. Rev. 2009, 30, 152–177.
- Kalupahana, N.S.; Moustaid-Moussa, N. The renin-angiotensin system: A link between obesity, inflammation and insulin resistance. Obes. Rev. 2012, 13, 136–149.
- Nandipati, K.C.; Subramanian, S.; Agrawal, D.K. Protein kinases: Mechanisms and downstream targets in inflammation-mediated obesity and insulin resistance. Mol. Cell. Biochem. 2017, 426, 27–45.
- Walke, P.B.; Bansode, S.B.; More, N.P.; Chaurasiya, A.H.; Joshi, R.S.; Kulkarni, M.J. Molecular investigation of glycated insulin-induced insulin resistance via insulin signaling and AGE-RAGE axis. Biochim. Biophys. Acta Mol. Basis Dis. 2021, 1867, 166029.
- Parwani, K.; Mandal, P. Role of advanced glycation end products and insulin resistance in diabetic nephropathy. Arch. Physiol. Biochem. 2020, 1–13.
- Jiang, Y.; Wang, Z.; Ma, B.; Fan, L.; Yi, N.; Lu, B.; Wang, Q.; Liu, R. GLP-1 improves adipocyte insulin sensitivity following induction of endoplasmic reticulum stress. Front. Pharmacol. 2018, 9, 1168.
- Sun, L.; Xie, C.; Wang, G.; Wu, Y.; Wu, Q.; Wang, X.; Liu, J.; Deng, Y.; Xia, J.; Chen, B.; et al. Gut microbiota and intestinal FXR mediate the clinical benefits of metformin. Nat. Med. 2018, 24, 1919–1929.
- Olefsky, J.M.; Glass, C.K. Macrophages, inflammation, and insulin resistance. Annu. Rev. Physiol. 2010, 72, 219–246.
- Daniele, G.; Guardado Mendoza, R.; Winnier, D.; Fiorentino, T.V.; Pengou, Z.; Cornell, J.; Andreozzi, F.; Jenkinson, C.; Cersosimo, E.; Federici, M.; et al. The inflammatory status score including IL-6, TNF-alpha, osteopontin, fractalkine, MCP-1 and adiponectin underlies whole-body insulin resistance and hyperglycemia in type 2 diabetes mellitus. Acta Diabetol. 2014, 51, 123–131.
- Palacios-Ortega, S.; Varela-Guruceaga, M.; Algarabel, M.; Ignacio Milagro, F.; Alfredo Martinez, J.; de Miguel, C. Effect of TNF-alpha on caveolin-1 expression and insulin signaling during adipocyte differentiation and in mature adipocytes. Cell. Physiol. Biochem. 2015, 36, 1499–1516.
- Hotamisligil, G.S.; Peraldi, P.; Budavari, A.; Ellis, R.; White, M.F.; Spiegelman, B.M. IRS-1-mediated inhibition of insulin receptor tyrosine kinase activity in TNF-alpha- and obesity-induced insulin resistance. Science 1996, 271, 665–668.
- Akash, M.S.; Shen, Q.; Rehman, K.; Chen, S. Interleukin-1 receptor antagonist: A new therapy for type 2 diabetes mellitus. J. Pharm. Sci. 2012, 101, 1647–1658.
- Wiegand, S.; Dannemann, A.; Krude, H.; Gruters, A. Impaired glucose tolerance and type 2 diabetes mellitus: A new field for pediatrics in Europe. Int. J. Obes. 2005, 29 (Suppl. 2), S136–S142.
- Bano, G. Glucose homeostasis, obesity and diabetes. Best Pract. Res. Clin. Obstet. Gynaecol. 2013, 27, 715–726.
- Sharma, R.; Tiwari, S. Renal gluconeogenesis in insulin resistance: A culprit for hyperglycemia in diabetes. World J. Diabetes 2021, 12, 556–568.
- Islam, R.; Kim, J.G.; Park, Y.; Cho, J.Y.; Cap, K.C.; Kho, A.R.; Chung, W.S.; Suh, S.W.; Park, J.B. Insulin induces phosphorylation of pyruvate dehydrogenase through RhoA activation pathway in HepG2 cells. FASEB J. 2019, 33, 2072–2083.
- Seckl, J.R.; Walker, B.R. Minireview: 11beta-hydroxysteroid dehydrogenase type 1- a tissue-specific amplifier of glucocorticoid action. Endocrinology 2001, 142, 1371–1376.
- Edwards, C.R.; Stewart, P.M.; Burt, D.; Brett, L.; McIntyre, M.A.; Sutanto, W.S.; de Kloet, E.R.; Monder, C. Localisation of 11 beta-hydroxysteroid dehydrogenase--tissue specific protector of the mineralocorticoid receptor. Lancet 1988, 2, 986–989.
- Alberts, P.; Nilsson, C.; Selen, G.; Engblom, L.O.; Edling, N.H.; Norling, S.; Klingstrom, G.; Larsson, C.; Forsgren, M.; Ashkzari, M.; et al. Selective inhibition of 11 beta-hydroxysteroid dehydrogenase type 1 improves hepatic insulin sensitivity in hyperglycemic mice strains. Endocrinology 2003, 144, 4755–4762.
- Yu, S.; Meng, S.; Xiang, M.; Ma, H. Phosphoenolpyruvate carboxykinase in cell metabolism: Roles and mechanisms beyond gluconeogenesis. Mol. Metab. 2021, 53, 101257.
- Liu, L.; Wang, Y.; Wang, J.; Dong, Y.; Chang, S.; Liu, X.; Lutfy, K.; Chen, H.; Friedman, T.C.; Jiang, M.; et al. Enhanced hexose-6-phosphate dehydrogenase expression in adipose tissue may contribute to diet-induced visceral adiposity. Int. J. Obes. 2018, 42, 1999–2011.
- Atanasov, A.G.; Odermatt, A. Readjusting the glucocorticoid balance: An opportunity for modulators of 11beta-hydroxysteroid dehydrogenase type 1 activity? Endocr. Metab. Immune Disord. Drug Targets 2007, 7, 125–140.
- Classen-Houben, D.; Schuster, D.; Da Cunha, T.; Odermatt, A.; Wolber, G.; Jordis, U.; Kueenburg, B. Selective inhibition of 11beta-hydroxysteroid dehydrogenase 1 by 18alpha-glycyrrhetinic acid but not 18beta-glycyrrhetinic acid. J. Steroid Biochem. Mol. Biol. 2009, 113, 248–252.
- Duan, C.; Liu, M.; Xu, H.; Tang, W.; Liu, J.; Hou, L.; Li, L. Decreased expression of GLUT4 in male CG-IUGR rats may play a vital role in their increased susceptibility to diabetes mellitus in adulthood. Acta Biochim. Biophys. Sin. 2016, 48, 872–882.
- Alam, F.; Islam, M.A.; Khalil, M.I.; Gan, S.H. Metabolic control of type 2 diabetes by targeting the GLUT4 glucose transporter: Intervention approaches. Curr. Pharm. Des. 2016, 22, 3034–3049.
- Wang, T.; Wang, J.; Hu, X.; Huang, X.J.; Chen, G.X. Current understanding of glucose transporter 4 expression and functional mechanisms. World J. Biol. Chem. 2020, 11, 76–98.
- Rani, R.; Dahiya, S.; Dhingra, D.; Dilbaghi, N.; Kim, K.H.; Kumar, S. Evaluation of anti-diabetic activity of glycyrrhizin-loaded nanoparticles in nicotinamide-streptozotocin-induced diabetic rats. Eur. J. Pharm. Sci. 2017, 106, 220–230.
- Rani, R.; Dahiya, S.; Dhingra, D.; Dilbaghi, N.; Kaushik, A.; Kim, K.H.; Kumar, S. Antidiabetic activity enhancement in streptozotocin + nicotinamide-induced diabetic rats through combinational polymeric nanoformulation. Int. J. Nanomed. 2019, 14, 4383–4395.
- Sherwani, S.I.; Khan, H.A.; Ekhzaimy, A.; Masood, A.; Sakharkar, M.K. Significance of HbA1c test in diagnosis and prognosis of diabetic patients. Biomark. Insights 2016, 11, 95–104.
- Negishi, M.; Irie, A.; Nagata, N.; Ichikawa, A. Specific binding of glycyrrhetinic acid to the rat liver membrane. Biochim. Biophys. Acta 1991, 1066, 77–82.
- Rastegari, A.; Mottaghitalab, F.; Dinarvand, R.; Amini, M.; Arefian, E.; Gholami, M.; Atyabi, F. Inhibiting hepatic gluconeogenesis by chitosan lactate nanoparticles containing CRTC2 siRNA targeted by poly(ethylene glycol)-glycyrrhetinic acid. Drug Deliv. Transl. Res. 2019, 9, 694–706.
- Adiels, M.; Olofsson, S.O.; Taskinen, M.R.; Boren, J. Overproduction of very low-density lipoproteins is the hallmark of the dyslipidemia in the metabolic syndrome. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 1225–1236.
- Savage, D.B.; Petersen, K.F.; Shulman, G.I. Disordered lipid metabolism and the pathogenesis of insulin resistance. Physiol. Rev. 2007, 87, 507–520.
- Eu, C.H.; Lim, W.Y.; Ton, S.H.; bin Abdul Kadir, K. Glycyrrhizic acid improved lipoprotein lipase expression, insulin sensitivity, serum lipid and lipid deposition in high-fat diet-induced obese rats. Lipids Health Dis. 2010, 9, 81.
- Sil, R.; Ray, D.; Chakraborti, A.S. Glycyrrhizin ameliorates metabolic syndrome-induced liver damage in experimental rat model. Mol. Cell. Biochem. 2015, 409, 177–189.
- Palacio Rojas, M.; Prieto, C.; Bermudez, V.; Garicano, C.; Nunez Nava, T.; Martinez, M.S.; Salazar, J.; Rojas, E.; Perez, A.; Marca Vicuna, P.; et al. Dyslipidemia: Genetics, lipoprotein lipase and HindIII polymorphism. F1000Research 2017, 6, 2073.
- Wung, S.F.; Kulkarni, M.V.; Pullinger, C.R.; Malloy, M.J.; Kane, J.P.; Aouizerat, B.E. The lipoprotein lipase gene in combined hyperlipidemia: Evidence of a protective allele depletion. Lipids Health Dis. 2006, 5, 19.
- Goldberg, I.J.; Eckel, R.H.; Abumrad, N.A. Regulation of fatty acid uptake into tissues: Lipoprotein lipase- and CD36-mediated pathways. J. Lipid Res. 2009, 50, S86–S90.
- Park, S.S.; Seo, Y.K. Excess accumulation of lipid impairs insulin sensitivity in skeletal muscle. Int. J. Mol. Sci. 2020, 21, 1949.
- Ferre, P.; Foufelle, F. SREBP-1c transcription factor and lipid homeostasis: Clinical perspective. Horm. Res. 2007, 68, 72–82.
- Yamamoto, M.; Nagasawa, Y.; Fujimori, K. Glycyrrhizic acid suppresses early stage of adipogenesis through repression of MEK/ERK-mediated C/EBPbeta and C/EBPdelta expression in 3T3-L1 cells. Chem.-Biol. Interact. 2021, 346, 109595.
- Moon, M.H.; Jeong, J.K.; Lee, Y.J.; Seol, J.W.; Ahn, D.C.; Kim, I.S.; Park, S.Y. 18beta-Glycyrrhetinic acid inhibits adipogenic differentiation and stimulates lipolysis. Biochem. Biophys. Res. Commun. 2012, 420, 805–810.
- Fu, Z.; Gilbert, E.R.; Liu, D. Regulation of insulin synthesis and secretion and pancreatic beta-cell dysfunction in diabetes. Curr. Diabetes Rev. 2013, 9, 25–53.
- Butler, A.E.; Janson, J.; Bonner-Weir, S.; Ritzel, R.; Rizza, R.A.; Butler, P.C. Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes 2003, 52, 102–110.
- Kubota, N.; Terauchi, Y.; Tobe, K.; Yano, W.; Suzuki, R.; Ueki, K.; Takamoto, I.; Satoh, H.; Maki, T.; Kubota, T.; et al. Insulin receptor substrate 2 plays a crucial role in beta cells and the hypothalamus. J. Clin. Investig. 2004, 114, 917–927.
- Fujimoto, K.; Polonsky, K.S. Pdx1 and other factors that regulate pancreatic beta-cell survival. Diabetes Obes. Metab. 2009, 11 (Suppl. 4), 30–37.
- Matschinsky, F.; Liang, Y.; Kesavan, P.; Wang, L.; Froguel, P.; Velho, G.; Cohen, D.; Permutt, M.A.; Tanizawa, Y.; Jetton, T.L.; et al. Glucokinase as pancreatic beta cell glucose sensor and diabetes gene. J. Clin. Investig. 1993, 92, 2092–2098.

