Type 2 diabetes mellitus (T2DM) is a chronic metabolic disease, which is characterized by hyperglycemia, chronic insulin resistance, progressive decline in β-cell function, and defect in insulin secretion. It has become one of the leading causes of death worldwide. There is no cure for T2DM, but it can be treated, and blood glucose levels can be controlled. It has been reported that diabetic patients may suffer from the adverse effects of conventional medicine. Therefore, alternative therapy, such as traditional Chinese medicine (TCM), can be used to manage and treat diabetes. Glycyrrhizic acid (GL) and its derivatives are suggested to be promising candidates for the treatment of T2DM and its complications. It is the principal bioactive constituent in licorice, one type of TCM.
- glycyrrhizic acid
- glycyrrhetinic acid
- ammonium glycyrrhizinate
- diammonium glycyrrhizinate
- type 2 diabetes mellitus
1. GL and Its Derivatives
Glycyrrhizic acid (GL) is an amphiphilic compound, its hydrophilic part is represented by the glucuronic acid residues, and its hydrophobic part is the glycyrrhetinic acid (GA) residue [1]. GA is not only a hydrolytic product of GL, but it is also one of the main bioactive components of licorice with two isoforms, 18α-GA and 18β-GA [2][3]. GL and its aglycon, GA, have promising therapeutic effects, which have attracted the researchers to develop them into clinical medicine, including anti-cancer, anti-inflammatory, anti-viral, anti-diabetic, and hepatoprotective effects [4][5]. However, studies found that the oral administration of GL is poorly absorbed from the gut, but it is slowly converted into 18β-GA under intestinal bacterial hydrolysis, and the hydrolysis product 18β-GA is absorbed in the gut [6][7]. Therefore, due to the poor bioavailability of GL, it has been designed to conjugate with various metals or ammonium to form water-soluble salts for improving its bioavailability, including ammonium glycyrrhizinate (AG), diammonium glycyrrhizinate (DG), and dipotassium glycyrrhizinate [8][9][10]. The structures of GL, GA, AG, and DG are shown in Figure 1.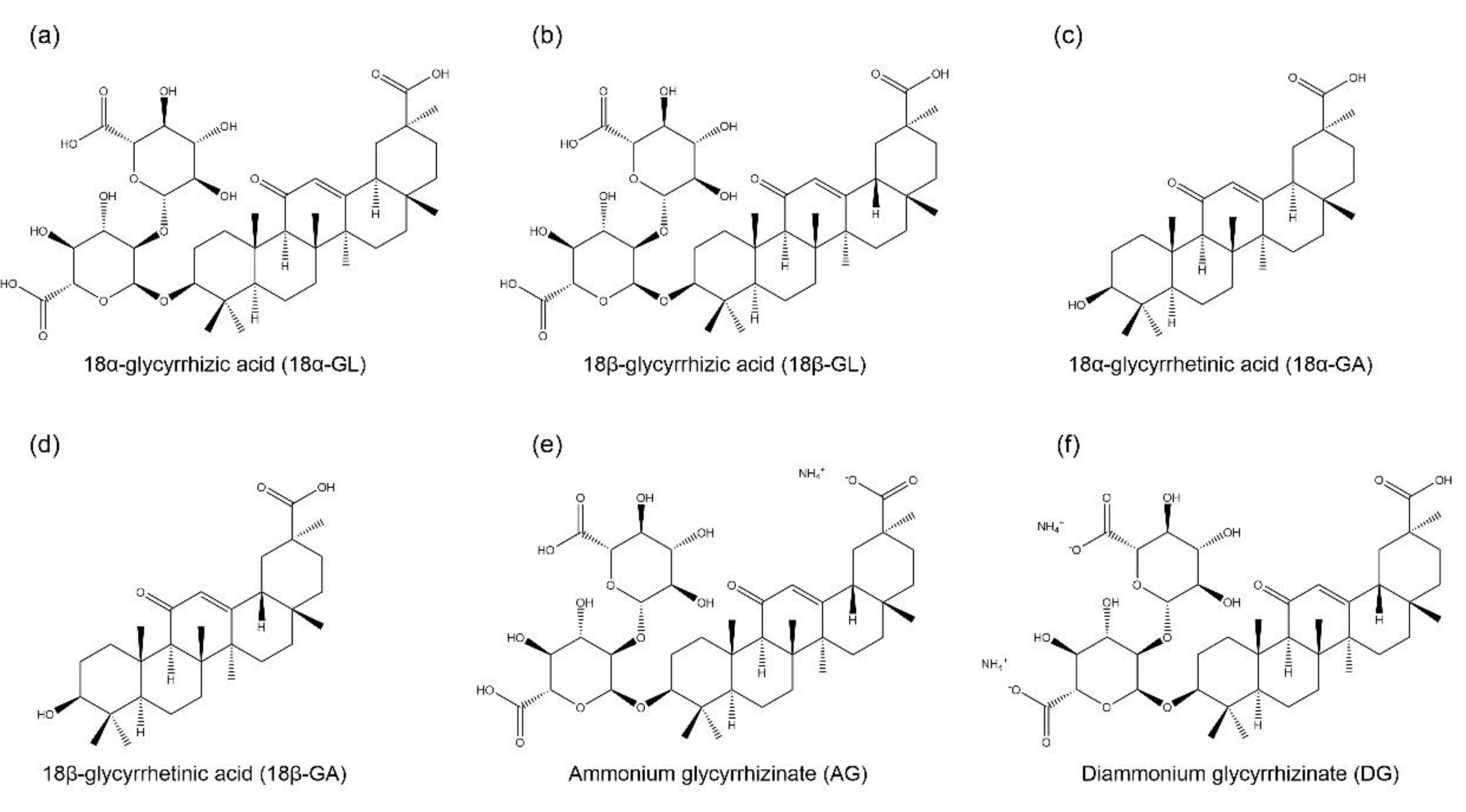
2. Anti-Diabetic Mechanisms of GL and Its Derivatives in T2DM
Type 2DM diabetes mellitus (T2DM) is mainly characterized by hyperglycemia, which eventually results in chronic insulin resistance in insulin-sensitive tissues and defective insulin secretion by pancreatic β-cells [11]. The anti-diabetic effects of GL and its derivatives have been confirmed in vitro and in vivo. The mechanisms include improving glucose tolerance [12] and insulin sensitivity [13], regulating glucose homeostasis [13] and lipid metabolism [14], and enhancing insulin secretion [15]. A schematic diagram shows the mechanisms of GL and its derivatives in improving insulin resistance in the liver and adipose tissue (Figure 2).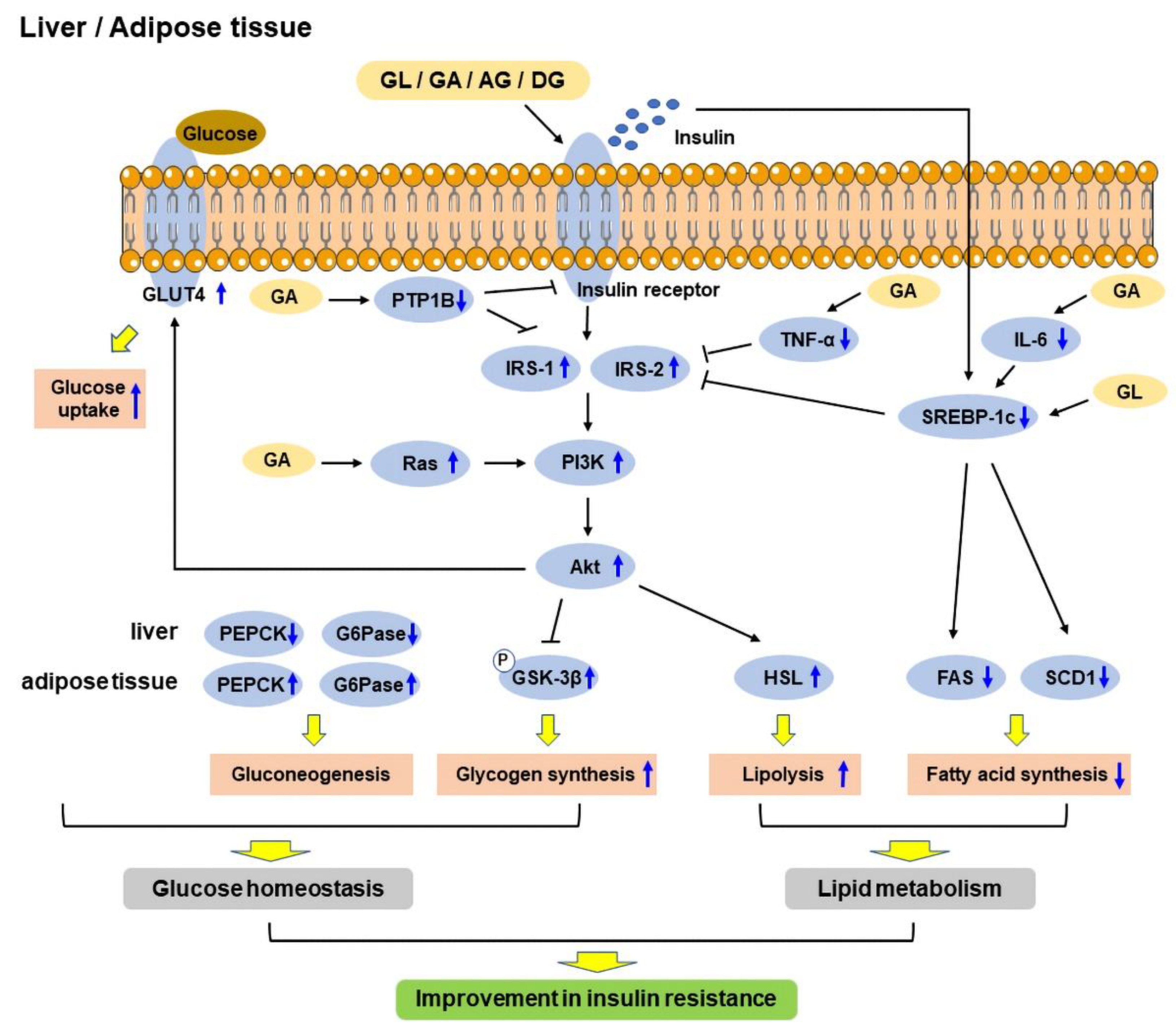
2.1. Insulin Resistance
Insulin is a hormone produced by the pancreas that controls glucose levels in the blood and helps in storing glucose in the insulin-sensitive organs, including skeletal muscle, fat, and the liver, where it is used to provide energy. The inability of responding to insulin in insulin-sensitive metabolic tissues, known as insulin resistance, is one of the main pathophysiology of T2DM, thus the tissues cannot uptake glucose from the blood easily. As a result, the pancreas produces more insulin to help in reducing blood glucose levels [16][17]. At a later stage, the pancreatic β-cells cannot compensate for the high demand of insulin; therefore, this eventually leads to β-cell dysfunction and defect in insulin secretion, and hyperglycemia occurs [18]. Studies have shown that GL, GA, and DG could improve insulin sensitivity in diabetic rodents [12][13][14][19][20][21][22][23][24][25][26][27][28][29][30]. There are several indicators of insulin resistance, including homeostasis model assessment of insulin resistance (HOMA-IR), homeostatic model assessment of β-cells (HOMA-β), and quantitative insulin sensitivity check index (QUICKI) [31]. HOMA was developed by Matthews et al., and it is used to quantify insulin resistance and β-cell function from fasting blood glucose and insulin levels [32], while QUICKI is another similar mathematical transformation from fasting blood glucose and insulin levels [33]. A study found that GL reduced HOMA-IR index [13][14][22][25][26][27][28], and increased HOMA-β and QUICKI indices [25] in diabetic rodents, indicating that GL could improve insulin sensitivity. Moreover, impaired fasting blood glucose levels is another indicator of insulin resistance and is characterized by a notable increase in hepatic insulin resistance [34]. GL and 18β-GA were demonstrated to reduce fasting blood glucose levels in diabetic rodents [12][13][14][21][22][25][27][28][29]. In addition, high fasting serum insulin levels is another sign of insulin resistance [35], and GL was found to decrease fasting serum insulin levels in diabetic rodents [12][13][14][28][29]. The insulin sensitivity of metabolic tissues plays a vital role in reducing blood glucose levels, and these tissues mainly include liver, adipose tissue, and skeletal muscle [36]. White adipose tissue (WAT) is an endocrine organ, which stores energy as triacylglycerols, and secretes hormones to regulate glucose homeostasis and adipokines to regulate inflammation. It was suggested that hypertrophy in WAT is associated with insulin resistance [37]. High-fat diet (HFD) was shown to increase visceral adipocyte size and induce adipose tissue inflammation in HFD-induced obese rats. In contrast, GL could decrease the size of adipocytes, thereby reducing body and WAT weights [19]. Moreover, it also reduced the adipocyte size and area in the subcutaneous WAT of rats [27][30]. Similarly, GL and DG could reduce the weights of WAT in HFD-induced obese rodents [19][23]. Furthermore, peroxisome proliferator-activated receptor-γ (PPAR-γ) is a nuclear receptor, which is highly expressed in adipose tissue [38]. The activation of PPAR-γ has been shown to upregulate the expressions of genes that are related to insulin signaling, thus enhancing insulin sensitivity [39]. Studies found that the GL treatment improved insulin sensitivity via upregulating the expression of PPAR-γ in the WAT and skeletal muscle [13][14][21]. On the other hand, the liver is responsible for glucose uptake and lipid synthesis and metabolism [40]. HFD increased liver weight, and GL reduced the liver weight and hepatic steatosis in HFD-induced diabetic mice [12]. Insulin receptor (IR) is a transmembrane receptor, which can be activated by insulin and insulin-like growth factor (IGF). IR and insulin receptor substrate (IRS) play a vital role in regulating glucose homeostasis in the pancreas and insulin-sensitive tissues [16]. IRS acts as a secondary messenger to transmit signals from insulin to downstream intracellular pathways, thus enhancing insulin sensitivity and the transcription of insulin-related genes [41]. GL was found to increase IR mRNA expression and its phosphorylation to enhance insulin sensitivity [19][20]. In addition, another study found that GL enhanced insulin sensitivity through the phosphorylation of IRS-1 and IRS-2 in HFD-induced diabetic mice [12]. Moreover, protein tyrosine phosphatase 1B (PTP1B), the main enzyme involved in IR desensitization, regulates insulin and leptin levels [42]. It is a negative regulator of insulin signaling pathway, and its inhibitors have become an attractive strategy to treat T2DM and obesity [43]. Interestingly, 18α-GA and 18β-GA were identified as competitive PTP1B inhibitors [44][45]. Two other derivatives of GA, namely, indole- and N-phenylpyrazole-GA, also showed potent non-competitive inhibition of PTP1B [46]. Insulin-like growth factor-1 (IGF-1) is a growth hormone that can enhance glucose uptake and reduce hepatic glucose production, thereby improving insulin sensitivity [47]. Insulin and IGF-1 regulate many signaling pathways, including Ras/mitogen-activated protein kinase (MAPK), and phosphoinositide 3-kinase (PI3K)/Akt pathway [16]. The activation of Ras is involved in the development of T2DM, and the inactivation of Ras genes improved insulin sensitivity [48]. In addition, PI3K/Akt pathway was identified as one of the vital pathways that are associated with insulin resistance [49]. GA was shown to improve glucose uptake and reverse insulin resistance by targeting Ras proteins and activating PI3K/Akt pathway, respectively [24]. Moreover, studies found that advanced glycation end products (AGE)-receptor for AGE (RAGE) axis activated oxidative stress and pro-inflammatory pathways, which contributed to insulin resistance [50][51]. GL was found to improve insulin resistance and downregulate RAGE expression spontaneously, suggesting that GL might improve insulin sensitivity via suppressing AGE-RAGE axis [22]. Glucagon-like peptide-1 (GLP-1), a 30-amino acid peptide hormone, is secreted by gut enteroendocrine cells, which improve insulin resistance in adipocytes [52]. It is currently used as a T2DM therapeutic. In addition, the inhibition of intestinal farnesoid X-activated receptor (FXR) or FXR deficiency was shown to promote GLP-1 secretion [53], and DG was also found to improve insulin resistance and glucose tolerance through downregulation of FXR [23]. Studies have shown that inflammation is associated with insulin resistance, and the most vital pro-inflammatory mediators include tumor necrosis factor-α (TNF-α) and interleukin 6 (IL-6). The inhibition of pro-inflammatory mediators was suggested to be one of the strategies for preventing the development of insulin resistance and the pathogenesis of diabetes [54][55][56][57][58]. GA was shown to reduce fasting plasma TNF-α and IL-6 levels to enhance insulin-responsive pathways [24].2.2. Glucose Tolerance and Homeostasis
Glucose tolerance is another indicator of diabetes and defined as the ability of the body to dispose glucose. Impaired glucose tolerance is often occurred in T2DM, and the progression from normal to impaired glucose tolerance is caused by insulin resistance [59]. Studies have shown that GL could improve glucose tolerance in diabetic rodents [12][13][29]. Glucose homeostasis can be affected by many factors, including insulin, glucagon, concentration of free fatty acids (FFA), and nutritional factors [60]. Gluconeogenesis is the process of glucose production, and gluconeogenic enzymes play a vital role in this process, including phosphoenolpyruvate carboxykinase (PEPCK), fructose-1,6-bisphosphatase (FBPase), and glucose-6-phosphatase (G6Pase) [61]. Glycogenesis is the process of glycogen synthesis, in which glucose is converted and stored as glycogen in the liver, which is activated by insulin in response to high blood glucose levels. Pyruvate dehydrogenase (PDase), an essential enzyme in glycogen synthesis, is also a substrate of glycogen synthase kinase 3β (GSK-3β), and GSK-3β induces pyruvate dehydrogenase E1α (PDH-E1α) phosphorylation in response to insulin to promote glycogen synthesis [62]. 11β-hydroxysteroid dehydrogenase (11β-HSD) is an enzyme involved in the pathogenesis of metabolic syndrome, which is characterized by hyperglycemia, dyslipidemia, diabetes, and arterial hypertension, and has two isoforms, 11β-HSD1 and 11β-HSD2 [63]. 11β-HSD1 is highly expressed in insulin-sensitive tissues, including liver, WAT, and skeletal muscle [64]. The inhibition of 11β-HSD1 ameliorated hyperglycemia and improved insulin sensitivity in diabetic mice [65]. In addition, PEPCK is a critical enzyme in gluconeogenesis, and the improved activity of PEPCK leads to elevated glucose output and aggravation of diabetes, whereas the defects of PEPCK result in lethal hypoglycemia [66]. Hexose-6-phosphate dehydrogenase (H6PDH), an enzyme that is localized in the endoplasmic reticulum lumen, converts glucose-6-phosphate (G6P) and nicotinamide adenine dinucleotide phosphate (NADP) to produce nicotinamide adenine dinucleotide phosphate hydrogen (NADPH). It provides a high [NADPH]/[NADP+] ratio for 11β-HSD1 amplification of intracellular active glucocorticoid production [67]. Moreover, increased 11β-HSD1-dependent active glucocorticoid production is associated with insulin resistance, T2DM, and its cardiovascular complications [68]. Studies found that GL could reduce PEPCK and G6Pase mRNA expressions to suppress gluconeogenesis, and upregulate PDase and GSK-3β mRNA expressions to increase glycogen synthesis in the liver [12][19][28]. Similarly, GL reduced PEPCK activities in the liver and kidney of normal rats [27], and alleviated the over-activities of PEPCK and G6Pase in the liver and kidney in rats with metabolic syndrome [14]. In addition, GL could reduce H6PDH activities in the liver, kidney, WAT, and muscle [27][28]. Moreover, GL could decrease the activities of 11β-HSD1 and 11β-HSD2 in the liver, kidney, WAT, and muscle to reduce blood glucose levels [26][27][28]. Furthermore, two isoforms of GA have different inhibition activities on 11β-HSD; 18α-GA selectively inhibits 11β-HSD1, but not 11β-HSD2, while 18β-GA favorably inhibits 11β-HSD2 [69]. Glucose transporter type 4 (GLUT4) is an insulin-regulated glucose transporter, which is involved in rapid glucose uptake in various kinds of cells to regulate glucose homeostasis, and its downregulation is closely associated with the development of glucose tolerance [70][71][72]. GA was found to promote GLUT4 expression by targeting Ras protein to regulate MAPK pathway [24]. Similarly, GL elevated GLUT4 expression in the skeletal muscle of HFD-induced diabetic rats to improve glucose homeostasis [13]. Moreover, GL and GA were shown to enhance insulin-stimulated glucose uptake in 3T3L-1 adipocytes [15]. Due to the poor bioavailability of GL, it has been formulated as nanoparticles. GL-loaded nanoparticles improved lipid profile and lowered fasting blood glucose levels in nicotinamide plus streptozotocin (STZ)-induced T2DM rats. Interestingly, the dosages used in GL-loaded nanoparticles were only a quarter of the dosages of the pure GL form [73][74]. Moreover, a combination of GL-loaded nanoparticles and thymoquinone-loaded nanocapsules was applied, displaying better anti-diabetic activities than when administered separately in nicotinamide and STZ-induced T2DM rats, including decreased blood glucose levels and improved lipid profile [74]. Glycosylated hemoglobin A1c is another biochemical parameter that is used to estimate the severity of diabetes, and its high level is an indicator of poor control of blood glucose levels [75]. Moreover, GL-loaded nanoparticles decreased the level of glycosylated hemoglobin A1c [73][74]. Furthermore, GA, as an efficient liver-specific ligand, was made to be a liver-targeted drug delivery carrier and conjugated with chitosan lactate and poly(ethylene glycol) to form nanoparticles containing siRNA-CREB regulated transcription coactivator 2 (CRTC2). These nanoparticles were shown to be accumulated in the liver after 2 h of treatment, reduce blood glucose levels, and inhibit hepatic gluconeogenesis in rats [76][77].2.3. Lipid Metabolism
The liver is responsible for the regulation of lipid metabolism, uptake of FFA, and export of fat [78]. In T2DM, lipid metabolism is altered, and fat uptake and delivery are imbalanced, which is linked with the over-production of very low-density lipoprotein (VLDL) [78][79]. Many studies found that HFD or high-fat-high-sucrose diet increased serum and hepatic triglyceride (TG), total cholesterol, low-density lipoprotein (LDL) cholesterol, and FFA levels, and decreased high-density lipoprotein (HDL) cholesterol levels in rodents [12][14][19][80][81]. In contrast, GL could improve serum lipid parameters, including decreases in serum and hepatic FFA, TG, total cholesterol, LDL cholesterol, and VLDL levels, but an increase in HDL cholesterol levels in diabetic rodents [12][14][19][30][73][74][80][81]. Similarly, GL-loaded nanoparticles could also decrease total cholesterol, TG, LDL, and VLDL levels, and increase HDL levels in diabetic rats [73]. Meanwhile, GL could also increase lipid uptake in cardiac and skeletal muscle [14]. Lipoprotein lipase (LPL) is a multi-functional glycoprotein enzyme, which plays a vital role in lipid parameters, such as the levels of TG, LDL cholesterol, HDL cholesterol, and VLDL, and its activity is upregulated by apolipoprotein C-II and downregulated by apolipoprotein C-III [82][83]. Increased LPL activity can be used as an indicator of enhanced lipid uptake into tissues [84]. GL was shown to upregulate LPL expression in the kidney, heart, muscle, and WAT [14][21][30][80]. In addition, other studies showed that GL promoted TG metabolism by inducing LPL activity, which upregulated hepatic apolipoprotein C-II expression and suppressed apolipoprotein C-III expression in HFD-induced diabetic mice [12]. Excess FFA and lipid droplets lead to intramuscular accumulation of lipid intermediates, which results in the inhibition of insulin signal transduction to impair insulin sensitivity [85]. GL was shown to decrease lipid deposition in the liver, kidney, heart, and muscle in HFD-induced diabetic rodents [12][80]. Interestingly, a study showed that GL treatment improved insulin sensitivity in HFD-induced obese rats, which might be through decreased lipid deposition [80]. Moreover, hepatic lipogenesis is regulated by a transcription factor, sterol regulatory element-binding transcription factor 1c (SREBP-1c), which is activated by insulin and regulates the genes that are involved in fatty acid and TG synthesis, including fatty acid synthetase (FAS) and stearoyl CoA desaturase (SCD) 1 [86]. GL was shown to improve lipid metabolism mainly through reducing hepatic lipogenesis and improving fatty acid metabolism, via downregulation of SREBP-1c, FAS, and SCD1 expressions, and upregulation of peroxisome proliferator-activated receptor-α (PPAR-α), carnitine palmitoyl transferase 1α (CPT1α), and acyl-coenzyme A dehydrogenase (ACADS), respectively [12]. GL and GA were also found to reduce the early stage of adipogenesis and lipid accumulation in 3T3-L1 adipocytes through downregulating CCAAT/enhancer-binding protein (C/EBP)-β and C/EBP-δ, PPAR-γ, and SCD expressions [15][87]. Similarly, 18β-GA was found to inhibit lipid accumulation, suppress adipogenic differentiation via inhibiting Akt phosphorylation and downregulating PPAR-γ and C/EBP-α expressions, and stimulate lipolysis via upregulation of adipose TG lipase (ATGL), hormone-sensitive lipase (HSL), and perilipin (Plin-1) expressions in 3T3-L1 adipocytes [88]. However, Yamamoto et al. showed that GL did not stimulate lipolysis in 3T3-L1 adipocytes [87].2.4. Insulin Secretion and Protection of Pancreatic β-Cells
Insulin is a hormone, which is produced by pancreatic β-cells in the islets of Langerhans, and it promotes glucose uptake from the blood into tissues. Insulin secretion is regulated by pancreatic β-cells in response to the increase in blood glucose levels [89]. In T2DM, chronic insulin resistance and a progressive decline in β-cell function result in β-cell dysfunction and defect in insulin secretion [90]. IRS-2 plays a vital role in the preservation of β-cell mass [91], while pancreas duodenum homeobox-1 (PDX-1) plays a role in β-cell survival and function [92]. In addition, glucokinase (GLK) acts as a glucose sensor to regulate insulin secretion [93]. GA was shown to enhance glucose-stimulated insulin secretion in isolated mouse islets, and upregulate IRS-2, PDX-1, and GLK expressions to improve β-cell viability [15]. A schematic diagram of GA-mediated glucose-stimulated insulin secretion in pancreatic β-cells is shown in Figure 3.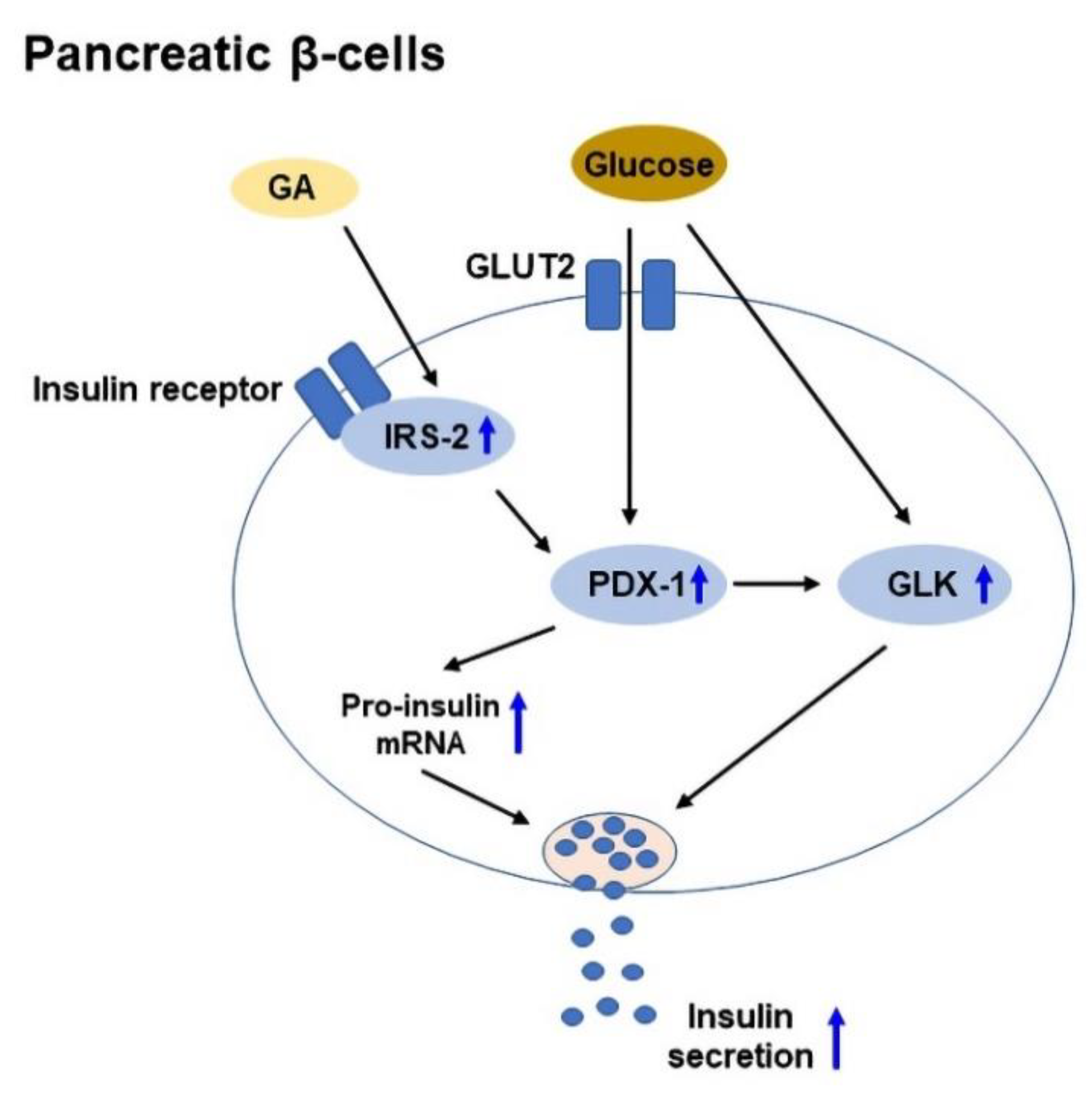
References
- Pastorino, G.; Cornara, L.; Soares, S.; Rodrigues, F.; Oliveira, M.B.P.P. Liquorice (Glycyrrhiza glabra): A phytochemical and pharmacological review. Phytother. Res. 2018, 32, 2323–2339.
- Newell-Price, J.D.C. Cushing Disease. In The Pituitary, 4th ed.; Melmed, S., Ed.; Academic Press: Cambridge, MA, USA, 2017; pp. 515–571.
- Sabbioni, C.; Mandrioli, R.; Ferranti, A.; Bugamelli, F.; Saracino, M.A.; Forti, G.C.; Fanali, S.; Raggi, M.A. Separation and analysis of glycyrrhizin, 18beta-glycyrrhetic acid and 18alpha-glycyrrhetic acid in liquorice roots by means of capillary zone electrophoresis. J. Chromatogr. A 2005, 1081, 65–71.
- Graebin, C.S. The Pharmacological Activities of Glycyrrhizinic Acid (“Glycyrrhizin”) and Glycyrrhetinic Acid. In Sweeteners: Pharmacology, Biotechnology, and Applications, 1st ed.; Merillon, J.-M., Ramawat, K.G., Eds.; Springer International Publishing: New York, NY, USA, 2017; pp. 245–261.
- Yang, L.; Jiang, Y.; Zhang, Z.; Hou, J.; Tian, S.; Liu, Y. The anti-diabetic activity of licorice, a widely used Chinese herb. J. Ethnopharmacol. 2020, 263, 113216.
- Takeda, S.; Ishthara, K.; Wakui, Y.; Amagaya, S.; Maruno, M.; Akao, T.; Kobashi, K. Bioavailability study of glycyrrhetic acid after oral administration of glycyrrhizin in rats; relevance to the intestinal bacterial hydrolysis. J. Pharm. Pharmacol. 1996, 48, 902–905.
- Akao, T.; Hayashi, T.; Kobashi, K.; Kanaoka, M.; Kato, H.; Kobayashi, M.; Takeda, S.; Oyama, T. Intestinal bacterial hydrolysis is indispensable to absorption of 18 beta-glycyrrhetic acid after oral administration of glycyrrhizin in rats. J. Pharm. Pharmacol. 1994, 46, 135–137.
- Yang, J.; Zhou, L.; Wang, J.; Wang, G.; Davey, A.K. The disposition of diammonium glycyrrhizinate and glycyrrhetinic acid in the isolated perfused rat intestine and liver. Planta Med. 2008, 74, 1351–1356.
- Jin, S.; Fu, S.; Han, J.; Jin, S.; Lv, Q.; Lu, Y.; Qi, J.; Wu, W.; Yuan, H. Improvement of oral bioavailability of glycyrrhizin by sodium deoxycholate/phospholipid-mixed nanomicelles. J. Drug Target. 2012, 20, 615–622.
- Fry, J.C. Natural low-calorie sweeteners. In Natural Food Additives, Ingredients and Flavourings, 1st ed.; Baines, D., Seal, R., Eds.; Woodhead Publishing: Sawston, UK, 2012; pp. 41–75.
- Galicia-Garcia, U.; Benito-Vicente, A.; Jebari, S.; Larrea-Sebal, A.; Siddiqi, H.; Uribe, K.B.; Ostolaza, H.; Martin, C. Pathophysiology of type 2 diabetes mellitus. Int. J. Mol. Sci. 2020, 21, 6275.
- Sun, X.; Duan, X.; Wang, C.; Liu, Z.; Sun, P.; Huo, X.; Ma, X.; Sun, H.; Liu, K.; Meng, Q. Protective effects of glycyrrhizic acid against non-alcoholic fatty liver disease in mice. Eur. J. Pharmacol. 2017, 806, 75–82.
- Sil, R.; Ray, D.; Chakraborti, A.S. Glycyrrhizin ameliorates insulin resistance, hyperglycemia, dyslipidemia and oxidative stress in fructose-induced metabolic syndrome-X in rat model. Indian J. Exp. Biol. 2013, 51, 129–138.
- Cheng, H.S.; Yaw, H.P.; Ton, S.H.; Choy, S.M.; Kong, J.M.; Abdul Kadir, K. Glycyrrhizic acid prevents high calorie diet-induced metabolic aberrations despite the suppression of peroxisome proliferator-activated receptor gamma expression. Nutrition 2016, 32, 995–1001.
- Ko, B.S.; Jang, J.S.; Hong, S.M.; Sung, S.R.; Lee, J.E.; Lee, M.Y.; Jeon, W.K.; Park, S. Changes in components, glycyrrhizin and glycyrrhetinic acid, in raw Glycyrrhiza uralensis Fisch, modify insulin sensitizing and insulinotropic actions. Biosci. Biotechnol. Biochem. 2007, 71, 1452–1461.
- Boucher, J.; Kleinridders, A.; Kahn, C.R. Insulin receptor signaling in normal and insulin-resistant states. Cold Spring Harb. Perspect. Biol. 2014, 6, a009191.
- National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK). Insulin Resistance & Prediabetes. Available online: https://www.niddk.nih.gov/health-information/diabetes/overview/what-is-diabetes/prediabetes-insulin-resistance (accessed on 12 June 2022).
- Fonseca, V.A. Defining and characterizing the progression of type 2 diabetes. Diabetes Care 2009, 32 (Suppl. 2), S151–S156.
- Abo El-Magd, N.F.; El-Mesery, M.; El-Karef, A.; El-Shishtawy, M.M. Glycyrrhizin ameliorates high fat diet-induced obesity in rats by activating NrF2 pathway. Life Sci. 2018, 193, 159–170.
- Liu, L.; Jiang, Y.; Steinle, J.J. Inhibition of HMGB1 protects the retina from ischemia-reperfusion, as well as reduces insulin resistance proteins. PLoS ONE 2017, 12, e0178236.
- Chia, Y.Y.; Ton, S.H.; Kadir, A.K. Effects of glycyrrhizic acid on peroxisome proliferator-activated receptor gamma (PPARgamma), lipoprotein lipase (LPL), serum lipid and HOMA-IR in rats. PPAR Res. 2010, 2010, 530265.
- Cheng, H.S.; Kong, J.M.; Ng, A.X.; Chan, W.K.; Ton, S.H.; Abdul Kadir, K. Novel inhibitory effects of glycyrrhizic acid on the accumulation of advanced glycation end product and its receptor expression. Nat. Prod. Bioprospect. 2014, 4, 325–333.
- Li, Y.; Hou, H.; Wang, X.; Dai, X.; Zhang, W.; Tang, Q.; Dong, Y.; Yan, C.; Wang, B.; Li, Z.; et al. Diammonium glycyrrhizinate ameliorates obesity through modulation of gut microbiota-conjugated BAs-FXR signaling. Front. Pharmacol. 2021, 12, 796590.
- Zhang, Y.; Yang, S.; Zhang, M.; Wang, Z.; He, X.; Hou, Y.; Bai, G. Glycyrrhetinic acid improves insulin-response pathway by regulating the balance between the Ras/MAPK and PI3K/Akt pathways. Nutrients 2019, 11, 604.
- Fatimah, M.; Malik, M.; Mushtaq, S.; Sarfraz, J.; Mushtaq, Z.; Chiragh, S. Dose dependent effect of glycyrrhizin on glycaemic control of type 2 diabetic rats. Khyber Med. Univ. J. 2020, 12, 121–125.
- Chia, Y.Y.; Ton, S.H.; Kadir, K.B. Effects of glycyrrhizic acid on 11 beta-hydroxysteroid dehydrogenase (11 betaHSD1 and 2) activities and HOMA-IR in rats at different treatment periods. Exp. Clin. Endocrinol. Diabetes 2010, 118, 617–624.
- Chia, Y.Y.; Liong, S.Y.; Ton, S.H.; Kadir, K.B. Amelioration of glucose homeostasis by glycyrrhizic acid through gluconeogenesis rate-limiting enzymes. Eur. J. Pharmacol. 2012, 677, 197–202.
- Chandramouli, C.; Ting, Y.S.; Lyn, L.Y.; Ha, T.S.; Kadir, K.A. Glycyrrhizic acid improves lipid and glucose metabolism in high-sucrose-fed rats. J. Endocrinol. Metab. 2011, 1, 125–141.
- Takii, H.; Kometani, T.; Nishimura, T.; Nakae, T.; Okada, S.; Fushiki, T. Antidiabetic effect of glycyrrhizin in genetically diabetic KK-Ay mice. Biol. Pharm. Bull. 2001, 24, 484–487.
- Lim, W.Y.; Chia, Y.Y.; Liong, S.Y.; Ton, S.H.; Kadir, K.A.; Husain, S.N. Lipoprotein lipase expression, serum lipid and tissue lipid deposition in orally-administered glycyrrhizic acid-treated rats. Lipids Health Dis. 2009, 8, 31.
- Muniyappa, R.; Madan, R.; Varghese, R.T. Assessing Insulin Sensitivity and Resistance in Humans. In Endotext; Feingold, K.R., Anawalt, B., Boyce, A., Chrousos, G., de Herder, W.W., Dhatariya, K., Dungan, K., Hershman, J.M., Hofland, J., Kalra, S., et al., Eds.; MD Text: South Dartmouth, MA, USA, 2021.
- Matthews, D.R.; Hosker, J.P.; Rudenski, A.S.; Naylor, B.A.; Treacher, D.F.; Turner, R.C. Homeostasis model assessment: Insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia 1985, 28, 412–419.
- Katsuki, A.; Sumida, Y.; Gabazza, E.C.; Murashima, S.; Urakawa, H.; Morioka, K.; Kitagawa, N.; Tanaka, T.; Araki-Sasaki, R.; Hori, Y.; et al. QUICKI is useful for following improvements in insulin sensitivity after therapy in patients with type 2 diabetes mellitus. J. Clin. Endocrinol. Metab. 2002, 87, 2906–2908.
- Sasaki, N.; Ozono, R.; Higashi, Y.; Maeda, R.; Kihara, Y. Association of insulin resistance, plasma glucose level, and serum insulin level with hypertension in a population with different stages of impaired glucose metabolism. J. Am. Heart Assoc. 2020, 9, e015546.
- Chaour, M.; Theroux, P.; Gilfix, B.M.; Campeau, L.; Lesperance, J.; Ghitescu, M.; Gelinas, F.; Solymoss, B.C. ‘True’ fasting serum insulin level, insulin resistance syndrome and coronary artery disease. Coron. Artery Dis. 1997, 8, 683–688.
- Zhang, H.; Zhang, C. Adipose “talks” to distant organs to regulate insulin sensitivity and vascular function. Obesity 2010, 18, 2071–2076.
- Hoffstedt, J.; Arner, E.; Wahrenberg, H.; Andersson, D.P.; Qvisth, V.; Lofgren, P.; Ryden, M.; Thorne, A.; Wiren, M.; Palmer, M.; et al. Regional impact of adipose tissue morphology on the metabolic profile in morbid obesity. Diabetologia 2010, 53, 2496–2503.
- Vidal-Puig, A.J.; Considine, R.V.; Jimenez-Linan, M.; Werman, A.; Pories, W.J.; Caro, J.F.; Flier, J.S. Peroxisome proliferator-activated receptor gene expression in human tissues. Effects of obesity, weight loss, and regulation by insulin and glucocorticoids. J. Clin. Investig. 1997, 99, 2416–2422.
- Leonardini, A.; Laviola, L.; Perrini, S.; Natalicchio, A.; Giorgino, F. Cross-talk between PPARgamma and insulin signaling and modulation of insulin sensitivity. PPAR Res. 2009, 2009, 818945.
- Santoleri, D.; Titchenell, P.M. Resolving the paradox of hepatic insulin resistance. Cell. Mol. Gastroenterol. Hepatol. 2019, 7, 447–456.
- Sesti, G.; Federici, M.; Hribal, M.L.; Lauro, D.; Sbraccia, P.; Lauro, R. Defects of the insulin receptor substrate (IRS) system in human metabolic disorders. FASEB J. 2001, 15, 2099–2111.
- Goldstein, B.J. Protein-tyrosine phosphatase 1B (PTP1B): A novel therapeutic target for type 2 diabetes mellitus, obesity and related states of insulin resistance. Curr. Drug Targets Immune Endocr. Metabol. Disord. 2001, 1, 265–275.
- Combs, A.P. Recent advances in the discovery of competitive protein tyrosine phosphatase 1B inhibitors for the treatment of diabetes, obesity, and cancer. J. Med. Chem. 2010, 53, 2333–2344.
- Seong, S.H.; Nguyen, D.H.; Wagle, A.; Woo, M.H.; Jung, H.A.; Choi, J.S. Experimental and computational study to reveal the potential of non-polar constituents from Hizikia fusiformis as dual protein tyrosine phosphatase 1B and alpha-glucosidase inhibitors. Mar. Drugs 2019, 17, 302.
- Na, M.; Cui, L.; Min, B.S.; Bae, K.; Yoo, J.K.; Kim, B.Y.; Oh, W.K.; Ahn, J.S. Protein tyrosine phosphatase 1B inhibitory activity of triterpenes isolated from Astilbe oreana. Bioorg. Med. Chem. Lett. 2006, 16, 3273–3276.
- De-la-Cruz-Martinez, L.; Duran-Becerra, C.; Gonzalez-Andrade, M.; Paez-Franco, J.C.; German-Acacio, J.M.; Espinosa-Chavez, J.; Torres-Valencia, J.M.; Perez-Villanueva, J.; Palacios-Espinosa, J.F.; Soria-Arteche, O.; et al. Indole- and pyrazole-glycyrrhetinic acid derivatives as PTP1B inhibitors: Synthesis, in vitro and in silico studies. Molecules 2021, 26, 4375.
- Moller, N.; Jorgensen, J.O. Effects of growth hormone on glucose, lipid, and protein metabolism in human subjects. Endocr. Rev. 2009, 30, 152–177.
- Kalupahana, N.S.; Moustaid-Moussa, N. The renin-angiotensin system: A link between obesity, inflammation and insulin resistance. Obes. Rev. 2012, 13, 136–149.
- Nandipati, K.C.; Subramanian, S.; Agrawal, D.K. Protein kinases: Mechanisms and downstream targets in inflammation-mediated obesity and insulin resistance. Mol. Cell. Biochem. 2017, 426, 27–45.
- Walke, P.B.; Bansode, S.B.; More, N.P.; Chaurasiya, A.H.; Joshi, R.S.; Kulkarni, M.J. Molecular investigation of glycated insulin-induced insulin resistance via insulin signaling and AGE-RAGE axis. Biochim. Biophys. Acta Mol. Basis Dis. 2021, 1867, 166029.
- Parwani, K.; Mandal, P. Role of advanced glycation end products and insulin resistance in diabetic nephropathy. Arch. Physiol. Biochem. 2020, 1–13.
- Jiang, Y.; Wang, Z.; Ma, B.; Fan, L.; Yi, N.; Lu, B.; Wang, Q.; Liu, R. GLP-1 improves adipocyte insulin sensitivity following induction of endoplasmic reticulum stress. Front. Pharmacol. 2018, 9, 1168.
- Sun, L.; Xie, C.; Wang, G.; Wu, Y.; Wu, Q.; Wang, X.; Liu, J.; Deng, Y.; Xia, J.; Chen, B.; et al. Gut microbiota and intestinal FXR mediate the clinical benefits of metformin. Nat. Med. 2018, 24, 1919–1929.
- Olefsky, J.M.; Glass, C.K. Macrophages, inflammation, and insulin resistance. Annu. Rev. Physiol. 2010, 72, 219–246.
- Daniele, G.; Guardado Mendoza, R.; Winnier, D.; Fiorentino, T.V.; Pengou, Z.; Cornell, J.; Andreozzi, F.; Jenkinson, C.; Cersosimo, E.; Federici, M.; et al. The inflammatory status score including IL-6, TNF-alpha, osteopontin, fractalkine, MCP-1 and adiponectin underlies whole-body insulin resistance and hyperglycemia in type 2 diabetes mellitus. Acta Diabetol. 2014, 51, 123–131.
- Palacios-Ortega, S.; Varela-Guruceaga, M.; Algarabel, M.; Ignacio Milagro, F.; Alfredo Martinez, J.; de Miguel, C. Effect of TNF-alpha on caveolin-1 expression and insulin signaling during adipocyte differentiation and in mature adipocytes. Cell. Physiol. Biochem. 2015, 36, 1499–1516.
- Hotamisligil, G.S.; Peraldi, P.; Budavari, A.; Ellis, R.; White, M.F.; Spiegelman, B.M. IRS-1-mediated inhibition of insulin receptor tyrosine kinase activity in TNF-alpha- and obesity-induced insulin resistance. Science 1996, 271, 665–668.
- Akash, M.S.; Shen, Q.; Rehman, K.; Chen, S. Interleukin-1 receptor antagonist: A new therapy for type 2 diabetes mellitus. J. Pharm. Sci. 2012, 101, 1647–1658.
- Wiegand, S.; Dannemann, A.; Krude, H.; Gruters, A. Impaired glucose tolerance and type 2 diabetes mellitus: A new field for pediatrics in Europe. Int. J. Obes. 2005, 29 (Suppl. 2), S136–S142.
- Bano, G. Glucose homeostasis, obesity and diabetes. Best Pract. Res. Clin. Obstet. Gynaecol. 2013, 27, 715–726.
- Sharma, R.; Tiwari, S. Renal gluconeogenesis in insulin resistance: A culprit for hyperglycemia in diabetes. World J. Diabetes 2021, 12, 556–568.
- Islam, R.; Kim, J.G.; Park, Y.; Cho, J.Y.; Cap, K.C.; Kho, A.R.; Chung, W.S.; Suh, S.W.; Park, J.B. Insulin induces phosphorylation of pyruvate dehydrogenase through RhoA activation pathway in HepG2 cells. FASEB J. 2019, 33, 2072–2083.
- Seckl, J.R.; Walker, B.R. Minireview: 11beta-hydroxysteroid dehydrogenase type 1- a tissue-specific amplifier of glucocorticoid action. Endocrinology 2001, 142, 1371–1376.
- Edwards, C.R.; Stewart, P.M.; Burt, D.; Brett, L.; McIntyre, M.A.; Sutanto, W.S.; de Kloet, E.R.; Monder, C. Localisation of 11 beta-hydroxysteroid dehydrogenase--tissue specific protector of the mineralocorticoid receptor. Lancet 1988, 2, 986–989.
- Alberts, P.; Nilsson, C.; Selen, G.; Engblom, L.O.; Edling, N.H.; Norling, S.; Klingstrom, G.; Larsson, C.; Forsgren, M.; Ashkzari, M.; et al. Selective inhibition of 11 beta-hydroxysteroid dehydrogenase type 1 improves hepatic insulin sensitivity in hyperglycemic mice strains. Endocrinology 2003, 144, 4755–4762.
- Yu, S.; Meng, S.; Xiang, M.; Ma, H. Phosphoenolpyruvate carboxykinase in cell metabolism: Roles and mechanisms beyond gluconeogenesis. Mol. Metab. 2021, 53, 101257.
- Liu, L.; Wang, Y.; Wang, J.; Dong, Y.; Chang, S.; Liu, X.; Lutfy, K.; Chen, H.; Friedman, T.C.; Jiang, M.; et al. Enhanced hexose-6-phosphate dehydrogenase expression in adipose tissue may contribute to diet-induced visceral adiposity. Int. J. Obes. 2018, 42, 1999–2011.
- Atanasov, A.G.; Odermatt, A. Readjusting the glucocorticoid balance: An opportunity for modulators of 11beta-hydroxysteroid dehydrogenase type 1 activity? Endocr. Metab. Immune Disord. Drug Targets 2007, 7, 125–140.
- Classen-Houben, D.; Schuster, D.; Da Cunha, T.; Odermatt, A.; Wolber, G.; Jordis, U.; Kueenburg, B. Selective inhibition of 11beta-hydroxysteroid dehydrogenase 1 by 18alpha-glycyrrhetinic acid but not 18beta-glycyrrhetinic acid. J. Steroid Biochem. Mol. Biol. 2009, 113, 248–252.
- Duan, C.; Liu, M.; Xu, H.; Tang, W.; Liu, J.; Hou, L.; Li, L. Decreased expression of GLUT4 in male CG-IUGR rats may play a vital role in their increased susceptibility to diabetes mellitus in adulthood. Acta Biochim. Biophys. Sin. 2016, 48, 872–882.
- Alam, F.; Islam, M.A.; Khalil, M.I.; Gan, S.H. Metabolic control of type 2 diabetes by targeting the GLUT4 glucose transporter: Intervention approaches. Curr. Pharm. Des. 2016, 22, 3034–3049.
- Wang, T.; Wang, J.; Hu, X.; Huang, X.J.; Chen, G.X. Current understanding of glucose transporter 4 expression and functional mechanisms. World J. Biol. Chem. 2020, 11, 76–98.
- Rani, R.; Dahiya, S.; Dhingra, D.; Dilbaghi, N.; Kim, K.H.; Kumar, S. Evaluation of anti-diabetic activity of glycyrrhizin-loaded nanoparticles in nicotinamide-streptozotocin-induced diabetic rats. Eur. J. Pharm. Sci. 2017, 106, 220–230.
- Rani, R.; Dahiya, S.; Dhingra, D.; Dilbaghi, N.; Kaushik, A.; Kim, K.H.; Kumar, S. Antidiabetic activity enhancement in streptozotocin + nicotinamide-induced diabetic rats through combinational polymeric nanoformulation. Int. J. Nanomed. 2019, 14, 4383–4395.
- Sherwani, S.I.; Khan, H.A.; Ekhzaimy, A.; Masood, A.; Sakharkar, M.K. Significance of HbA1c test in diagnosis and prognosis of diabetic patients. Biomark. Insights 2016, 11, 95–104.
- Negishi, M.; Irie, A.; Nagata, N.; Ichikawa, A. Specific binding of glycyrrhetinic acid to the rat liver membrane. Biochim. Biophys. Acta 1991, 1066, 77–82.
- Rastegari, A.; Mottaghitalab, F.; Dinarvand, R.; Amini, M.; Arefian, E.; Gholami, M.; Atyabi, F. Inhibiting hepatic gluconeogenesis by chitosan lactate nanoparticles containing CRTC2 siRNA targeted by poly(ethylene glycol)-glycyrrhetinic acid. Drug Deliv. Transl. Res. 2019, 9, 694–706.
- Adiels, M.; Olofsson, S.O.; Taskinen, M.R.; Boren, J. Overproduction of very low-density lipoproteins is the hallmark of the dyslipidemia in the metabolic syndrome. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 1225–1236.
- Savage, D.B.; Petersen, K.F.; Shulman, G.I. Disordered lipid metabolism and the pathogenesis of insulin resistance. Physiol. Rev. 2007, 87, 507–520.
- Eu, C.H.; Lim, W.Y.; Ton, S.H.; bin Abdul Kadir, K. Glycyrrhizic acid improved lipoprotein lipase expression, insulin sensitivity, serum lipid and lipid deposition in high-fat diet-induced obese rats. Lipids Health Dis. 2010, 9, 81.
- Sil, R.; Ray, D.; Chakraborti, A.S. Glycyrrhizin ameliorates metabolic syndrome-induced liver damage in experimental rat model. Mol. Cell. Biochem. 2015, 409, 177–189.
- Palacio Rojas, M.; Prieto, C.; Bermudez, V.; Garicano, C.; Nunez Nava, T.; Martinez, M.S.; Salazar, J.; Rojas, E.; Perez, A.; Marca Vicuna, P.; et al. Dyslipidemia: Genetics, lipoprotein lipase and HindIII polymorphism. F1000Research 2017, 6, 2073.
- Wung, S.F.; Kulkarni, M.V.; Pullinger, C.R.; Malloy, M.J.; Kane, J.P.; Aouizerat, B.E. The lipoprotein lipase gene in combined hyperlipidemia: Evidence of a protective allele depletion. Lipids Health Dis. 2006, 5, 19.
- Goldberg, I.J.; Eckel, R.H.; Abumrad, N.A. Regulation of fatty acid uptake into tissues: Lipoprotein lipase- and CD36-mediated pathways. J. Lipid Res. 2009, 50, S86–S90.
- Park, S.S.; Seo, Y.K. Excess accumulation of lipid impairs insulin sensitivity in skeletal muscle. Int. J. Mol. Sci. 2020, 21, 1949.
- Ferre, P.; Foufelle, F. SREBP-1c transcription factor and lipid homeostasis: Clinical perspective. Horm. Res. 2007, 68, 72–82.
- Yamamoto, M.; Nagasawa, Y.; Fujimori, K. Glycyrrhizic acid suppresses early stage of adipogenesis through repression of MEK/ERK-mediated C/EBPbeta and C/EBPdelta expression in 3T3-L1 cells. Chem.-Biol. Interact. 2021, 346, 109595.
- Moon, M.H.; Jeong, J.K.; Lee, Y.J.; Seol, J.W.; Ahn, D.C.; Kim, I.S.; Park, S.Y. 18beta-Glycyrrhetinic acid inhibits adipogenic differentiation and stimulates lipolysis. Biochem. Biophys. Res. Commun. 2012, 420, 805–810.
- Fu, Z.; Gilbert, E.R.; Liu, D. Regulation of insulin synthesis and secretion and pancreatic beta-cell dysfunction in diabetes. Curr. Diabetes Rev. 2013, 9, 25–53.
- Butler, A.E.; Janson, J.; Bonner-Weir, S.; Ritzel, R.; Rizza, R.A.; Butler, P.C. Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes 2003, 52, 102–110.
- Kubota, N.; Terauchi, Y.; Tobe, K.; Yano, W.; Suzuki, R.; Ueki, K.; Takamoto, I.; Satoh, H.; Maki, T.; Kubota, T.; et al. Insulin receptor substrate 2 plays a crucial role in beta cells and the hypothalamus. J. Clin. Investig. 2004, 114, 917–927.
- Fujimoto, K.; Polonsky, K.S. Pdx1 and other factors that regulate pancreatic beta-cell survival. Diabetes Obes. Metab. 2009, 11 (Suppl. 4), 30–37.
- Matschinsky, F.; Liang, Y.; Kesavan, P.; Wang, L.; Froguel, P.; Velho, G.; Cohen, D.; Permutt, M.A.; Tanizawa, Y.; Jetton, T.L.; et al. Glucokinase as pancreatic beta cell glucose sensor and diabetes gene. J. Clin. Investig. 1993, 92, 2092–2098.