 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Yasmin Boyle | -- | 3625 | 2022-10-07 06:53:57 | | | |
| 2 | Lindsay Dong | -8 word(s) | 3617 | 2022-10-08 07:31:48 | | |
Video Upload Options
Potassium ion channels are a class of transmembrane proteins involved in maintaining the electrical microenvironment of cells. In cancers, these proteins are known to play a significant role in the development of cancer hallmarks like proliferation, invasion, and adaptive drug resistance. Malignant central nervous system (CNS) cancers are notoriously difficult to treat, with just one-third of patients surviving five years post-diagnosis. Their location within the brain and brainstem presents several challenges to successful treatment, including surgical inaccessibility and designing effective therapies that pass the brain’s protective barrier. Furthermore, high-grade CNS cancer is also prone to recurrence, and the secondary tumours that arise are highly resistant to treatment.
1. Introduction
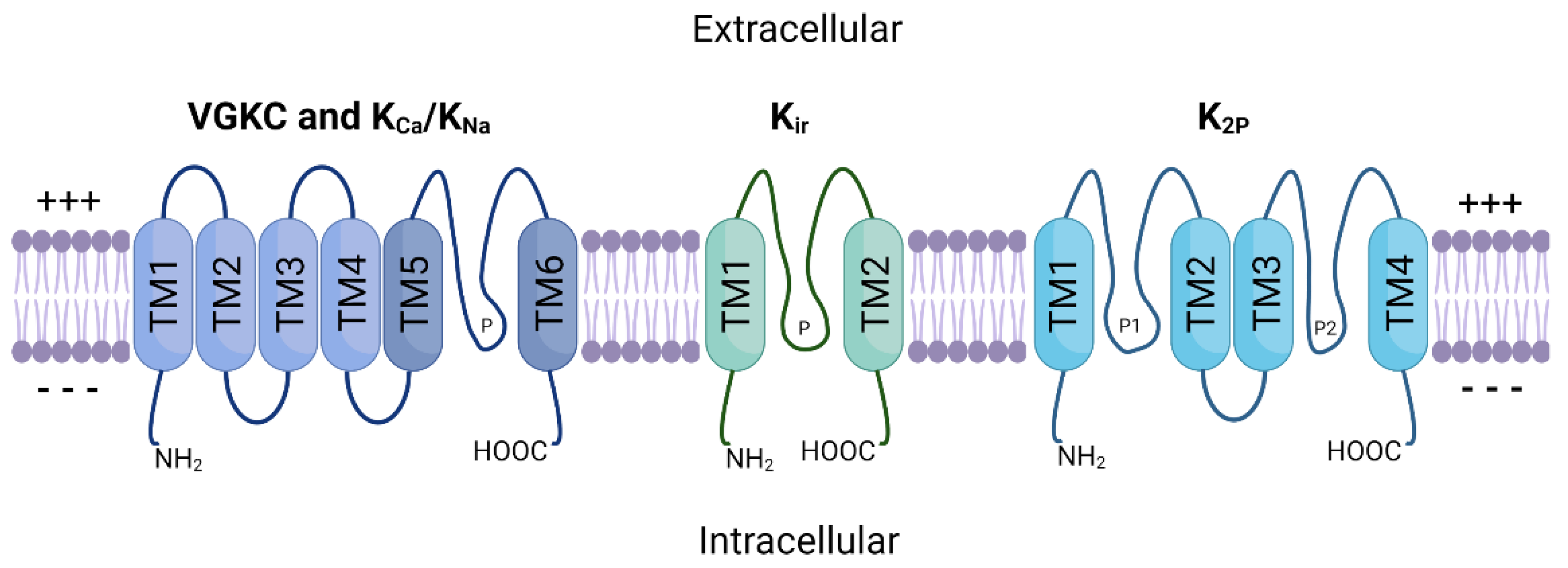
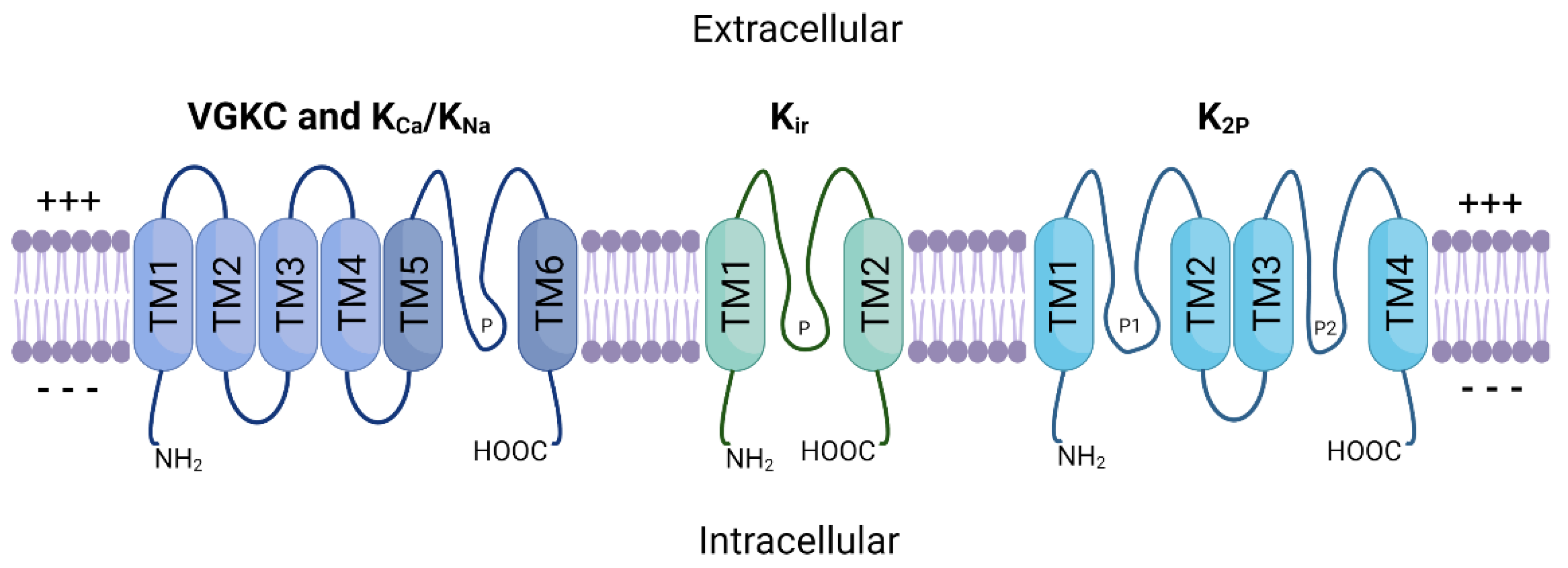
2. Potassium Ion Channels
3. Malignant CNS Cancer
3.1. Targeted Therapies
3.2. Cellular Plasticity and Drug Resistance
4. Potassium Ion Channels in Malignant CNS Cancer
4.1. High-Grade Glioma
4.2. Low-Grade Glioma
4.3. Medulloblastoma
5. Potassium Ion Channels as Therapeutic Targets
6. Conclusions
References
- Gabashvili, I.S.; Sokolowski, B.H.A.; Morton, C.C.; Giersch, A.B.S. Ion Channel Gene Expression in the Inner Ear. J. Assoc. Res. Otolaryngol. 2007, 8, 305–328.
- Tian, C.; Zhu, R.; Zhu, L.; Qiu, T.; Cao, Z.; Kang, T. Potassium Channels: Structures, Diseases, and Modulators. Chem. Biol. Drug Des. 2014, 83, 1–26.
- Alberts, B.; Johnson, A.; Lewis, J.; Raff, M.; Roberts, K.; Walter, P. Ion Channels and the Electrical Properties of Membranes. In Molecular Biology of the Cell, 4th ed.; Garland Science: New York, NY, USA, 2002.
- Chrysafides, S.M.; Bordes, S.; Sharma, S. Physiology, Resting Potential. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022.
- Deemyad, T.; Lüthi, J.; Spruston, N. Astrocytes Integrate and Drive Action Potential Firing in Inhibitory Subnetworks. Nat. Commun. 2018, 9, 4336.
- Grant, A.O. Cardiac Ion Channels. Circ. Arrhythm. Electrophysiol. 2009, 2, 185–194.
- Zavodnik, I.B.; Piasecka, A.; Szosland, K.; Bryszewska, M. Human Red Blood Cell Membrane Potential and Fluidity in Glucose Solutions. Scand. J. Clin. Lab. Investig. 1997, 57, 59–63.
- Bordey, A.; Sontheimer, H. Electrophysiological Properties of Human Astrocytic Tumor Cells In Situ: Enigma of Spiking Glial Cells. J. Neurophysiol. 1998, 79, 2782–2793.
- Kim, J.-B. Channelopathies. Korean J. Pediatr. 2014, 57, 1–18.
- Prevarskaya, N.; Skryma, R.; Shuba, Y. Ion Channels and the Hallmarks of Cancer. Trends Mol. Med. 2010, 16, 107–121.
- Hanahan, D.; Weinberg, R.A. The Hallmarks of Cancer. Cell 2000, 100, 57–70.
- Jackson, W.F. Potassium Channels in Regulation of Vascular Smooth Muscle Contraction and Growth. Adv. Pharmacol. 2017, 78, 89–144.
- Neylon, C.B.; Lang, R.J.; Fu, Y.; Bobik, A.; Reinhart, P.H. Molecular Cloning and Characterization of the Intermediate-Conductance Ca2+-Activated K+ Channel in Vascular Smooth Muscle: Relationship between K(Ca) Channel Diversity and Smooth Muscle Cell Function. Circ. Res. 1999, 85, e33–e43.
- Bi, D.; Toyama, K.; Lemaître, V.; Takai, J.; Fan, F.; Jenkins, D.P.; Wulff, H.; Gutterman, D.D.; Park, F.; Miura, H. The Intermediate Conductance Calcium-Activated Potassium Channel KCa3.1 Regulates Vascular Smooth Muscle Cell Proliferation via Controlling Calcium-Dependent Signaling. J. Biol. Chem. 2013, 288, 15843–15853.
- Kuang, Q.; Purhonen, P.; Hebert, H. Structure of Potassium Channels. Cell. Mol. Life Sci. 2015, 72, 3677–3693.
- Sun, X.; Zaydman, M.A.; Cui, J. Regulation of Voltage-Activated K+ Channel Gating by Transmembrane β Subunits. Front. Pharmacol. 2012, 3, 63.
- Jiang, Y.; Lee, A.; Chen, J.; Ruta, V.; Cadene, M.; Chait, B.T.; MacKinnon, R. X-ray Structure of a Voltage-Dependent K+ Channel. Nature 2003, 423, 33–41.
- Isacoff, E.Y.; Jan, L.Y.; Minor, D.L. Conduits of Life’s Spark: A Perspective on Ion Channel Research since the Birth of Neuron. Neuron 2013, 80, 658–674.
- Kaczmarek, L.K. Slack, Slick and Sodium-Activated Potassium Channels. ISRN Neurosci. 2013, 2013, 354262.
- Huang, X.; Jan, L.Y. Targeting Potassium Channels in Cancer. J. Cell Biol. 2014, 206, 151–162.
- Doyle, D.A.; Cabral, J.M.; Pfuetzner, R.A.; Kuo, A.; Gulbis, J.M.; Cohen, S.L.; Chait, B.T.; MacKinnon, R. The Structure of the Potassium Channel: Molecular Basis of K+ Conduction and Selectivity. Science 1998, 280, 69–77.
- Rasmusson, R.L.; Morales, M.J.; Wang, S.; Liu, S.; Campbell, D.L.; Brahmajothi, M.V.; Strauss, H.C. Inactivation of Voltage-Gated Cardiac K+ Channels. Circ. Res. 1998, 82, 739–750.
- Sigworth, F.J. Voltage Gating of Ion Channels. Q. Rev. Biophys. 1994, 27, 1–40.
- Sabater, V.G.; Rigby, M.; Burrone, J. Voltage-Gated Potassium Channels Ensure Action Potential Shape Fidelity in Distal Axons. J. Neurosci. 2021, 41, 5372–5385.
- Blackiston, D.J.; McLaughlin, K.A.; Levin, M. Bioelectric Controls of Cell Proliferation. Cell Cycle 2009, 8, 3519–3528.
- He, K.; McCord, M.C.; Hartnett, K.A.; Aizenman, E. Regulation of Pro-Apoptotic Phosphorylation of Kv2.1 K+ Channels. PLoS ONE 2015, 10, e0129498.
- Wang, Q.; Curran, M.E.; Splawski, I.; Burn, T.C.; Millholland, J.M.; VanRaay, T.J.; Shen, J.; Timothy, K.W.; Vincent, G.M.; de Jager, T.; et al. Positional Cloning of a Novel Potassium Channel Gene: KVLQT1 Mutations Cause Cardiac Arrhythmias. Nat. Genet. 1996, 12, 17–23.
- Kshatri, A.S.; Gonzalez-Hernandez, A.; Giraldez, T. Physiological Roles and Therapeutic Potential of Ca2+ Activated Potassium Channels in the Nervous System. Front. Mol. Neurosci. 2018, 11, 258.
- Sweet, T.-B.; Cox, D.H. Measuring the Influence of the BKCa β1 Subunit on Ca2+ Binding to the BKCa Channel. J. Gen. Physiol. 2009, 133, 139–150.
- Lee, U.S.; Cui, J. BK Channel Activation: Structural and Functional Insights. Trends Neurosci. 2010, 33, 415–423.
- Hager, N.A.; McAtee, C.K.; Lesko, M.A.; O’Donnell, A.F. Inwardly Rectifying Potassium Channel Kir2.1 and Its “Kir-Ious” Regulation by Protein Trafficking and Roles in Development and Disease. Front. Cell Dev. Biol. 2021, 9, 796136.
- Belus, M.T.; Rogers, M.A.; Elzubeir, A.; Josey, M.; Rose, S.; Andreeva, V.; Yelick, P.C.; Bates, E.A. Kir2.1 Is Important for Efficient BMP Signaling in Mammalian Face Development. Dev. Biol. 2018, 444 (Suppl. 1), S297–S307.
- Feliciangeli, S.; Chatelain, F.C.; Bichet, D.; Lesage, F. The Family of K2P Channels: Salient Structural and Functional Properties. J. Physiol. 2015, 593, 2587–2603.
- Zúñiga, L.; Zúñiga, R. Understanding the Cap Structure in K2P Channels. Front. Physiol. 2016, 7, 228.
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249.
- Miller, K.D.; Ostrom, Q.T.; Kruchko, C.; Patil, N.; Tihan, T.; Cioffi, G.; Fuchs, H.E.; Waite, K.A.; Jemal, A.; Siegel, R.L.; et al. Brain and Other Central Nervous System Tumor Statistics, 2021. CA Cancer J. Clin. 2021, 71, 381–406.
- Liu, Y.; Tyler, E.; Lustick, M.; Klein, D.; Walter, K.A. Healthcare Costs for High-Grade Glioma. Anticancer Res. 2019, 39, 1375–1381.
- Stupp, R.; Brada, M.; van den Bent, M.J.; Tonn, J.-C.; Pentheroudakis, G.; ESMO Guidelines Working Group. High-Grade Glioma: ESMO Clinical Practice Guidelines for Diagnosis, Treatment and Follow-Up. Ann. Oncol. 2014, 25 (Suppl. 3), iii93–iii101.
- Yao, M.; Li, S.; Wu, X.; Diao, S.; Zhang, G.; He, H.; Bian, L.; Lu, Y. Cellular Origin of Glioblastoma and Its Implication in Precision Therapy. Cell. Mol. Immunol. 2018, 15, 737–739.
- Wang, Y.; Jiang, T. Understanding High Grade Glioma: Molecular Mechanism, Therapy and Comprehensive Management. Cancer Lett. 2013, 331, 139–146.
- Shergalis, A.; Bankhead, A.; Luesakul, U.; Muangsin, N.; Neamati, N. Current Challenges and Opportunities in Treating Glioblastoma. Pharmacol. Rev. 2018, 70, 412–445.
- Chen, R.; Cohen, A.L.; Colman, H. Targeted Therapeutics in Patients with High-Grade Gliomas: Past, Present, and Future. Curr. Treat. Options Oncol. 2016, 17, 42.
- An, Z.; Aksoy, O.; Zheng, T.; Fan, Q.-W.; Weiss, W.A. Epidermal Growth Factor Receptor and EGFRvIII in Glioblastoma: Signaling Pathways and Targeted Therapies. Oncogene 2018, 37, 1561–1575.
- Swartz, A.M.; Li, Q.-J.; Sampson, J.H. Rindopepimut: A Promising Immunotherapeutic for the Treatment of Glioblastoma Multiforme. Immunotherapy 2014, 6, 679–690.
- Inman, S. Rindopepimut Misses OS Endpoint in Phase III Glioblastoma Trial. Available online: https://www.onclive.com/view/rindopepimut-misses-os-endpoint-in-phase-iii-glioblastoma-trial (accessed on 11 June 2022).
- Batchelor, T.T.; Mulholland, P.; Neyns, B.; Nabors, L.B.; Campone, M.; Wick, A.; Mason, W.; Mikkelsen, T.; Phuphanich, S.; Ashby, L.S.; et al. Phase III Randomized Trial Comparing the Efficacy of Cediranib as Monotherapy, and in Combination with Lomustine, versus Lomustine Alone in Patients with Recurrent Glioblastoma. J. Clin. Oncol. 2013, 31, 3212–3218.
- Gilbert, M.R.; Dignam, J.J.; Armstrong, T.S.; Wefel, J.S.; Blumenthal, D.T.; Vogelbaum, M.A.; Colman, H.; Chakravarti, A.; Pugh, S.; Won, M.; et al. A Randomized Trial of Bevacizumab for Newly Diagnosed Glioblastoma. N. Engl. J. Med. 2014, 370, 699–708.
- Chinot, O.L.; Wick, W.; Mason, W.; Henriksson, R.; Saran, F.; Nishikawa, R.; Carpentier, A.F.; Hoang-Xuan, K.; Kavan, P.; Cernea, D.; et al. Bevacizumab plus Radiotherapy-Temozolomide for Newly Diagnosed Glioblastoma. N. Engl. J. Med. 2014, 370, 709–722.
- Neftel, C.; Laffy, J.; Filbin, M.G.; Hara, T.; Shore, M.E.; Rahme, G.J.; Richman, A.R.; Silverbush, D.; Shaw, M.L.; Hebert, C.M.; et al. An Integrative Model of Cellular States, Plasticity, and Genetics for Glioblastoma. Cell 2019, 178, 835–849.
- Debanne, D.; Daoudal, G.; Sourdet, V.; Russier, M. Brain Plasticity and Ion Channels. J. Physiol. Paris 2003, 97, 403–414.
- Liebau, S.; Kleger, A.; Levin, M.; Yu, S.P. Stem Cells and Ion Channels. Stem Cells Int. 2013, 2013, 238635.
- Sturm, D.; Witt, H.; Hovestadt, V.; Khuong-Quang, D.-A.; Jones, D.T.W.; Konermann, C.; Pfaff, E.; Tönjes, M.; Sill, M.; Bender, S.; et al. Hotspot Mutations in H3F3A and IDH1 Define Distinct Epigenetic and Biological Subgroups of Glioblastoma. Cancer Cell 2012, 22, 425–437.
- Greenall, S.A.; Donoghue, J.F.; Van Sinderen, M.; Dubljevic, V.; Budiman, S.; Devlin, M.; Street, I.; Adams, T.E.; Johns, T.G. EGFRvIII-Mediated Transactivation of Receptor Tyrosine Kinases in Glioma: Mechanism and Therapeutic Implications. Oncogene 2015, 34, 5277–5287.
- Pillay, V.; Allaf, L.; Wilding, A.L.; Donoghue, J.F.; Court, N.W.; Greenall, S.A.; Scott, A.M.; Johns, T.G. The Plasticity of Oncogene Addiction: Implications for Targeted Therapies Directed to Receptor Tyrosine Kinases. Neoplasia 2009, 11, 448–458.
- Friedmann-Morvinski, D. Glioblastoma Heterogeneity and Cancer Cell Plasticity. Crit. Rev. Oncog. 2014, 19, 327–336.
- Osuka, S.; Meir, E.G.V. Overcoming Therapeutic Resistance in Glioblastoma: The Way Forward. J. Clin. Investig. 2017, 127, 415–426.
- Lathia, J.D.; Mack, S.C.; Mulkearns-Hubert, E.E.; Valentim, C.L.L.; Rich, J.N. Cancer Stem Cells in Glioblastoma. Genes Dev. 2015, 29, 1203–1217.
- Hadjipanayis, C.G.; Van Meir, E.G. Tumor Initiating Cells in Malignant Gliomas: Biology and Implications for Therapy. J. Mol. Med. 2009, 87, 363–374.
- Singh, N.; Miner, A.; Hennis, L.; Mittal, S. Mechanisms of Temozolomide Resistance in Glioblastoma—A Comprehensive Review. Cancer Drug Resist. 2021, 4, 17–43.
- Kim, H.; Zheng, S.; Amini, S.S.; Virk, S.M.; Mikkelsen, T.; Brat, D.J.; Grimsby, J.; Sougnez, C.; Muller, F.; Hu, J.; et al. Whole-Genome and Multisector Exome Sequencing of Primary and Post-Treatment Glioblastoma Reveals Patterns of Tumor Evolution. Genome Res. 2015, 25, 316–327.
- Orzan, F.; De Bacco, F.; Crisafulli, G.; Pellegatta, S.; Mussolin, B.; Siravegna, G.; D’Ambrosio, A.; Comoglio, P.M.; Finocchiaro, G.; Boccaccio, C. Genetic Evolution of Glioblastoma Stem-Like Cells from Primary to Recurrent Tumor. Stem Cells 2017, 35, 2218–2228.
- Zhang, Y.; Dube, C.; Gibert, M.; Cruickshanks, N.; Wang, B.; Coughlan, M.; Yang, Y.; Setiady, I.; Deveau, C.; Saoud, K.; et al. The P53 Pathway in Glioblastoma. Cancers 2018, 10, E297.
- Ruggieri, P.; Mangino, G.; Fioretti, B.; Catacuzzeno, L.; Puca, R.; Ponti, D.; Miscusi, M.; Franciolini, F.; Ragona, G.; Calogero, A. The Inhibition of KCa3.1 Channels Activity Reduces Cell Motility in Glioblastoma Derived Cancer Stem Cells. PLoS ONE 2012, 7, e47825.
- Ocasio, J.K.; Babcock, B.; Malawsky, D.; Weir, S.J.; Loo, L.; Simon, J.M.; Zylka, M.J.; Hwang, D.; Dismuke, T.; Sokolsky, M.; et al. ScRNA-Seq in Medulloblastoma Shows Cellular Heterogeneity and Lineage Expansion Support Resistance to SHH Inhibitor Therapy. Nat. Commun. 2019, 10, 5829.
- Vanner, R.J.; Remke, M.; Gallo, M.; Selvadurai, H.J.; Coutinho, F.; Lee, L.; Kushida, M.; Head, R.; Morrissy, S.; Zhu, X.; et al. Quiescent Sox2+ Cells Drive Hierarchical Growth and Relapse in Sonic Hedgehog Subgroup Medulloblastoma. Cancer Cell 2014, 26, 33–47.
- Mansouri, S.; Nejad, R.; Karabork, M.; Ekinci, C.; Solaroglu, I.; Aldape, K.D.; Zadeh, G. Sox2: Regulation of Expression and Contribution to Brain Tumors. CNS Oncol. 2016, 5, 159–173.
- Ransom, C.B.; Sontheimer, H. BK Channels in Human Glioma Cells. J. Neurophysiol. 2001, 85, 790–803.
- Wondergem, R.; Bartley, J.W. Menthol Increases Human Glioblastoma Intracellular Ca2+, BK Channel Activity and Cell Migration. J. Biomed. Sci. 2009, 16, 90.
- Abdullaev, I.F.; Rudkouskaya, A.; Mongin, A.A.; Kuo, Y.-H. Calcium-Activated Potassium Channels BK and IK1 Are Functionally Expressed in Human Gliomas but Do Not Regulate Cell Proliferation. PLoS ONE 2010, 5, e12304.
- Weaver, A.K.; Liu, X.; Sontheimer, H. Role for Calcium-Activated Potassium Channels (BK) in Growth Control of Human Malignant Glioma Cells. J. Neurosci. Res. 2004, 78, 224–234.
- Kraft, R.; Krause, P.; Jung, S.; Basrai, D.; Liebmann, L.; Bolz, J.; Patt, S. BK Channel Openers Inhibit Migration of Human Glioma Cells. Pflug. Arch. 2003, 446, 248–255.
- Newman, E.A. Inward-Rectifying Potassium Channels in Retinal Glial (Müller) Cells. J. Neurosci. 1993, 13, 3333–3345.
- Olsen, M.L.; Sontheimer, H. Mislocalization of Kir Channels in Malignant Glia. Glia 2004, 46, 63–73.
- Traynelis, S.F.; Dingledine, R. Potassium-Induced Spontaneous Electrographic Seizures in the Rat Hippocampal Slice. J. Neurophysiol. 1988, 59, 259–276.
- Augustus, M.; Pineau, D.; Aimond, F.; Azar, S.; Lecca, D.; Scamps, F.; Muxel, S.; Darlix, A.; Ritchie, W.; Gozé, C.; et al. Identification of CRYAB+ KCNN3+ SOX9+ Astrocyte-Like and EGFR+ PDGFRA+ OLIG1+ Oligodendrocyte-Like Tumoral Cells in Diffuse IDH1-Mutant Gliomas and Implication of NOTCH1 Signalling in Their Genesis. Cancers 2021, 13, 2107.
- Huang, X.; Dubuc, A.M.; Hashizume, R.; Berg, J.; He, Y.; Wang, J.; Chiang, C.; Cooper, M.K.; Northcott, P.A.; Taylor, M.D.; et al. Voltage-Gated Potassium Channel EAG2 Controls Mitotic Entry and Tumor Growth in Medulloblastoma via Regulating Cell Volume Dynamics. Genes Dev. 2012, 26, 1780–1796.
- Huang, X.; He, Y.; Dubuc, A.M.; Hashizume, R.; Zhang, W.; Reimand, J.; Yang, H.; Wang, T.A.; Stehbens, S.J.; Younger, S.; et al. EAG2 Potassium Channel with Evolutionarily Conserved Function as a Brain Tumor Target. Nat. Neurosci. 2015, 18, 1236–1246.
- Francisco, M.A.; Wanggou, S.; Fan, J.J.; Dong, W.; Chen, X.; Momin, A.; Abeysundara, N.; Min, H.-K.; Chan, J.; McAdam, R.; et al. Chloride Intracellular Channel 1 Cooperates with Potassium Channel EAG2 to Promote Medulloblastoma Growth. J. Exp. Med. 2020, 217, e20190971.
- Fan, J.; Pusong, R.; Morrissy, S.; Farooq, H.; Taylor, M.; Huang, X. MEDU-28. Eliminating the Root of Medulloblastoma by Targeting a Voltage-Gated Potassium Channel. Neuro Oncol. 2019, 21, ii109.
- Santos, R.; Ursu, O.; Gaulton, A.; Bento, A.P.; Donadi, R.S.; Bologa, C.G.; Karlsson, A.; Al-Lazikani, B.; Hersey, A.; Oprea, T.I.; et al. A Comprehensive Map of Molecular Drug Targets. Nat. Rev. Drug Discov. 2017, 16, 19–34.
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A Major Update to the DrugBank Database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082.
- Vandenberg, J.I.; Perry, M.D.; Perrin, M.J.; Mann, S.A.; Ke, Y.; Hill, A.P. HERG K+ Channels: Structure, Function, and Clinical Significance. Physiol. Rev. 2012, 92, 1393–1478.
- Brendorp, B.; Pedersen, O.; Torp-Pedersen, C.; Sahebzadah, N.; Køber, L. A Benefit-Risk Assessment of Class III Antiarrhythmic Agents. Drug Saf. 2002, 25, 847–865.
- Guardado-Mendoza, R.; Prioletta, A.; Jiménez-Ceja, L.M.; Sosale, A.; Folli, F. The Role of Nateglinide and Repaglinide, Derivatives of Meglitinide, in the Treatment of Type 2 Diabetes Mellitus. Arch. Med. Sci. 2013, 9, 936–943.

