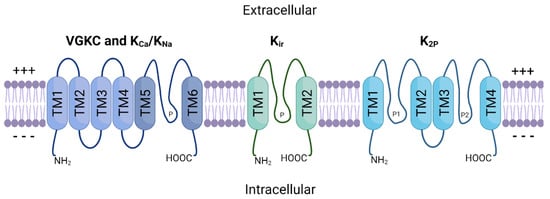
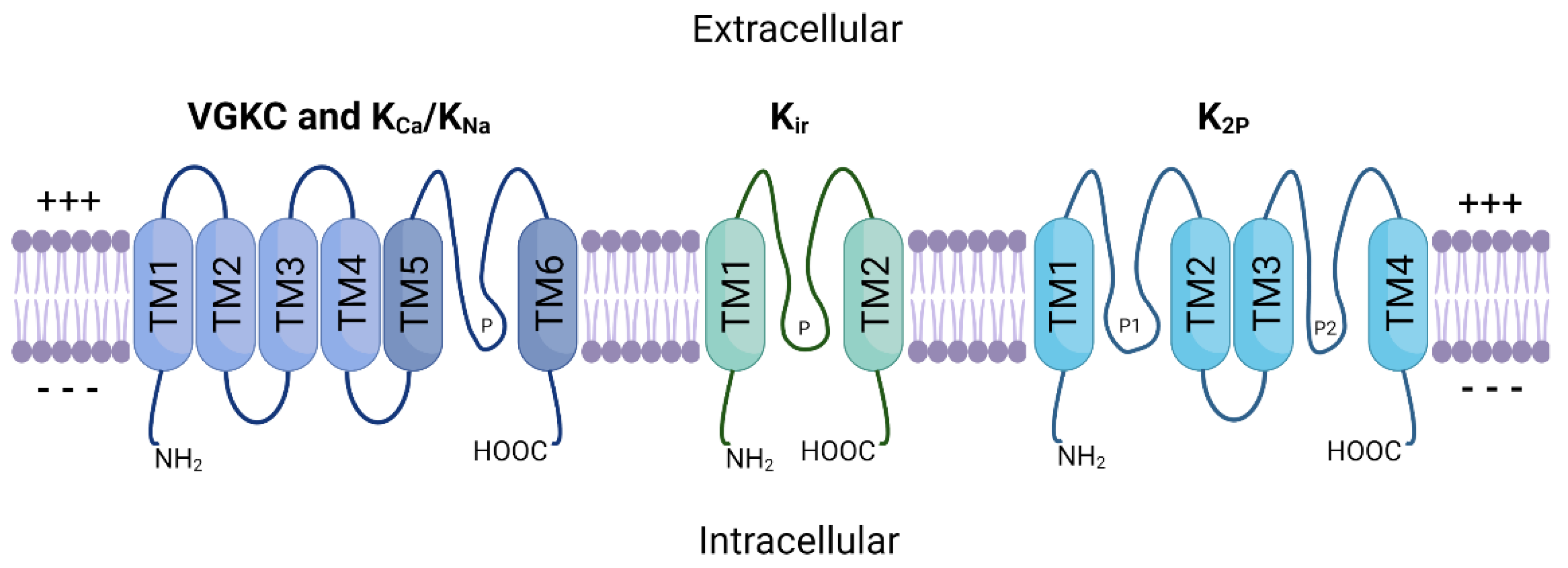
3.2. Cellular Plasticity and Drug Resistance
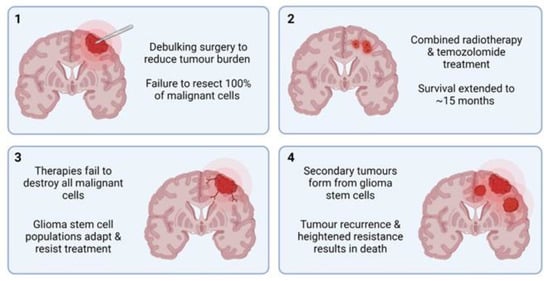
Both healthy and malignant neural cells are highly plastic, a significant barrier preventing the success of targeted HGG therapy. Sturm et al.
[52][141] identified the six distinct states, differentiated by their DNA methylation patterns, between which HGG cells continually and reversibly transition. This plasticity underpins adaptive drug resistance, in which alternative signalling pathways are activated in response to the targeted blockade of integral proteins
[53][54][55][142,143,144]. This process largely relies on glioma stem cells (GSCs), unique malignant cell populations capable of forming heterogenous HGG tumours. Many cancerous cells that remain following standard HGG treatment can be classified as GSCs. They are characterised by their heightened resistance to drugs, unlimited proliferation, and multipotent differentiation
[56][145].
GSCs are believed to be responsible for HGG’s adaptive response to treatment by developing alternative oncogenic pathways that bypass the targeted proteins or pathways
[57][58][148,149]. For example, TMZ resistance is common in GBM tumours, and GSCs are believed to be primarily responsible for this
[59](reviewed in [150]). Firstly, whole genome sequencing of matched primary and recurrent GBM tumours showed common deleterious mutations in
TP53 that originated from GSC populations
[60][61][151,152]. Mutations in
TP53 are common in recurrent GBM, and linked to heightened invasion, migration, proliferation and GSC propagation
[62][153].
It has been established that ion channels, including potassium ion channels, are key proteins in stem cells, including GSCs. Following treatment with the K
Ca3.1 antagonist TRAM-34, GSC cell mobility and migration was significantly reduced
[63][155].
Similarly, to glioma, MB tumours are largely heterogenous and diverse, exhibiting various cell types, including stem cell-like populations (MBSCs)
[64][158]. Single-cell analysis of MB tumours following vismodegib treatment revealed an increase in the population of MBSCs, which express the oncogene,
SOX2 [65][159].
SOX2 plays a significant role in cancer cell stemness and is often overexpressed in the stem-cell populations of both HGG and MB
[66][160].
4. Potassium Ion Channels in Malignant CNS Cancer
4.1. High-Grade Glioma
The K
+-regulating ion channel family is perhaps the most well understood regarding HGG pathophysiology and tumourigenesis, although several published studies have conflicting results. For example, BK channel overexpression has been identified in biopsy samples of GBM patient tumours, with the level of overexpression directly related to increasing malignancy grade
[67][163]. The importance of BK channels was further solidified by the discovery of a novel splice isoform of the protein (gBK channels) that is exclusively expressed in GBM. The gBK channels appear to be directly involved in the cellular volume/size changes required for invasion of healthy brain parenchyma. Increased cytosolic Ca
2+ concentration, for example via treatment with menthol, activates gBK channels and facilitates greater GBM cell migration and invasion
[68][164]. While siRNA-induced knockdown of gBK channels in GBM does not change proliferation rates
[69][165], various BK channel inhibitors can decrease cellular proliferation and increase tumour shrinkage in GBM cell cultures
[70][166]. Conversely, BK channel activators were shown to reduce glioma cell migration by as much as 50%, suggesting that increased BK channel activity may, in fact, reduce glioma invasiveness
[71][167].
Inwardly rectifying K+ (K
ir) channels have also been implicated in GBM progression and tumourigenesis. K
ir channels are found in abundance in normal glial cells, where they produce large, inwardly rectifying K
+ currents that stabilise the membrane potential to approximately −90 mV
[72][168]. However, when glial cells become malignant, the cellular membrane becomes depolarised, increasing up to −20 mV
[8][9]. Further studies showed that K
ir channels are expressed in GBM but are localised to the nucleus rather than the plasma membrane
[73][169]. This reduces the effect that K
ir channel function can have on overall cellular membrane potential. It may also contribute to epileptic seizures in many patients with GBM, as mislocalisation of K
ir proteins can result in increased extracellular K+ ion concentration, which is associated with spontaneous seizures
[74][170]. Thus, although K
ir channels may not be as directly related to tumourigenesis as some other ion channel subtypes, they may represent potential targets in treating GBM-induced seizures.
4.2. Low-Grade Glioma
Compared to HGG, there is significantly less information available regarding how potassium ion channels contribute to LGG disease progression. However, a few studies have explored how specific potassium ion channel subtypes are expressed in LGG. Diffuse grade II
IDH1-mutant gliomas are particularly rare, and often affect young adults. These tumours are particularly heterogenous, displaying multiple different cell subtypes within a single tumour, but the underlying mechanisms of this were not well understood. Augustus et al.
[75][182] identified that multiple proteins were involved in developing this intratumoural diversity, including K
Ca2.3. In the astrocyte-like tumoural cell populations, electrically active K
Ca2.3 channels were highly expressed, while the oligodendrocyte-like tumour cells showed significantly lower expression. This suggests that K
Ca2.3 may have potential to be used as a marker for astrocyte-like tumoural cells, aiding in patient tumour classification and therapeutic strategies.
4.3. Medulloblastoma
The majority of existing studies targeted at potassium ion channels and medulloblastoma have centred around the K
v11.1 subtype. Huang et al.
[76][106] were the first to observe that K
v11.1 was overexpressed in MB tumours samples derived from various subgroups compared with healthy brain. K
v11.1 knockdown reduced tumour cell growth in vitro and reduced tumour burden in xenograft mouse models. K
v11.1 was confined inside the cell before the G2 phase of the cell cycle, and upon reaching late G2, it was trafficked to the plasma membrane. Therefore, K
v11.1 knockdown induced G2 arrest, resulting in mitotic catastrophe and cell death. Further studies established that K
v11.1 was specifically enriched in the most invasive subpopulations of MB cells, where it regulated changes to cell shape and volume, thereby facilitating migration
[77][186]. This process is facilitated by the chloride ion channel, CLIC1, during which both proteins aggregate at lipid rafts to regulate cell volume
[78][187]. Simultaneous knockdown K
v11.1 and CLIC1 resulted in the suppressed growth/tumour burden in human MB cell lines and fruit fly models. Similarly, loss of the VGKC subtype K
v2.2 results in significantly improved survival in mouse models of MB
[79][188].
5. Potassium Ion Channels as Therapeutic Targets
Ion channels represent one of the most common targets of currently approved drugs, second only to G-coupled protein receptors
[80][190]. Several potassium ion channel antagonists, categorised as class III antiarrhythmic agents, have been developed to treat cardiac arrhythmias. These include dofetilide, amiodarone, sotalol, and ibutilide, which act on the K
v11.1, 11.2, and 11.3 VGKCs, respectively, but have known off-target actions other proteins
[81][191]. It is well established that K
v11 channels are responsible for mediating the heart’s inwardly rectifying K
+ current. Furthermore, missense mutations in these channels can result in type 2 long QT syndrome, triggering arrhythmias
[82][192]. Due to their integral role in maintaining K
+ homeostasis in the heart, these drugs have potential serious adverse effects and can cause proarrhythmia
[83][193].
Not all ion channel drugs block channel function. A small group of FDA-approved drugs work by forcing the channel to remain open, allowing increased K
+ ion flow. Most of these drugs, including minoxidil (targets K
ir1.1) and diazoxide (targets K
ir6.2), are classed as vasodilators and are used to regulate blood pressure, while ezogabine targets K
v7.2 to 7.5 and acts as an anti-convulsant
[81][191]. Alteration of VGKC function via blockade or sustained opening of the channel is of clinical value across several conditions. This is promising regarding CNS cancer treatment because these malignancies appear to be reliant on VGKC function for both growth and metastasis. Furthermore, because many potassium ion channel antagonists are already approved for clinical use in humans, there is potential for rapid clinical translation via drug repurposing. This includes the class III antiarrhythmics, as well as nateglinide and repaglinide, used in the management of diabetes
[84][194]. When an existing drug is repurposed for a new function, such as cancer treatment, the process involved in receiving FDA approval for the new indication/s is vastly shorter than it would be for an entirely new molecule. With further research, there is potential for developing novel drugs that target specific potassium ion channel subtypes in several CNS cancers.
6. Conclusions
Thanks to the collaborative efforts of numerous research teams,
theour understanding of the roles of ion channels in tumourigenesis has grown substantially in recent decades. The discovery that ion channels act as crucial drivers of homeostasis via proliferation and cell cycle regulation, and not just as propagators of action potentials, provided the basis from which much of
theour understanding has grown. It is now understood that ion channels are integral to the processes underlying cellular plasticity, the primary hurdle preventing the successful and curative treatment of HGG. Malignant HGG cells repurpose the endogenously expressed ion channels in healthy glial cells to further their development. Potassium ion channels are perhaps the most well-studied ion channel class in HGG oncogenesis, with many individual studies demonstrating how these cells utilise VGKC, K
Ca, and K
ir channels for enhanced growth and metastasis. Several VGKC-targeting drugs have already received FDA approval, proving that alteration of their normal activity is possible without causing unwanted off-target effects.
Further research should aim to elucidate the roles that potassium ion channels play in CNS cancers and to understand the cellular pathways and downstream proteins they influence. siRNA or CRISPR-mediated knockout studies involving key potassium ion channel subtypes in GBM cell lines would provide further insight into their involvement in key malignant cellular processes. Electrophysiological studies could also help determine whether or not these channels act independently of their current-modifying ion channel functions. Pharmacological inhibition of specific subtypes using repurposed drugs may also demonstrate the potential therapeutic value of these channels in GBM treatment. Finally, the development and use of accurate in vivo models of highly malignant tumours would be of great value, as studies involving cell lines alone can be somewhat limited in scope and potential.