Potassium ion channels are a class of transmembrane proteins involved in maintaining the electrical microenvironment of cells. In cancers, these proteins are known to play a significant role in the development of cancer hallmarks like proliferation, invasion, and adaptive drug resistance. Malignant central nervous system (CNS) cancers are notoriously difficult to treat, with just one-third of patients surviving five years post-diagnosis. Their location within the brain and brainstem presents several challenges to successful treatment, including surgical inaccessibility and designing effective therapies that pass the brain’s protective barrier. Furthermore, high-grade CNS cancer is also prone to recurrence, and the secondary tumours that arise are highly resistant to treatment.
- potassium ion channels
- high grade glioma
- glioblastoma
- medulloblastoma
1. Introduction
2. Potassium Ion Channels
The potassium ion channel family is the largest of all ion channel classes and comprises four subfamilies: voltage-gated (VGKC), sodium and calcium-activated (KNa, KCa), inward-rectifying (Kir), and two-pore domain (K2P) potassium channels [30][15]. The channels in each subfamily differ based on their domain structure, gating mechanisms and function. Figure 1 provides a schematic representation of the general structure of these protein subfamilies. VGKC, KNa and KCa channels share a basic 4 α-subunit structure with a single pore-forming region (TM5–TM6), while Kir channels consist of only two α-subunits. K2P channels possess two separate pore-forming domains and a four α-subunit structure. Furthermore, these multimeric proteins are often associated with one or more auxiliary β-subunits (encoded by genes KCNE1–5) that further influence channel gating [31][16].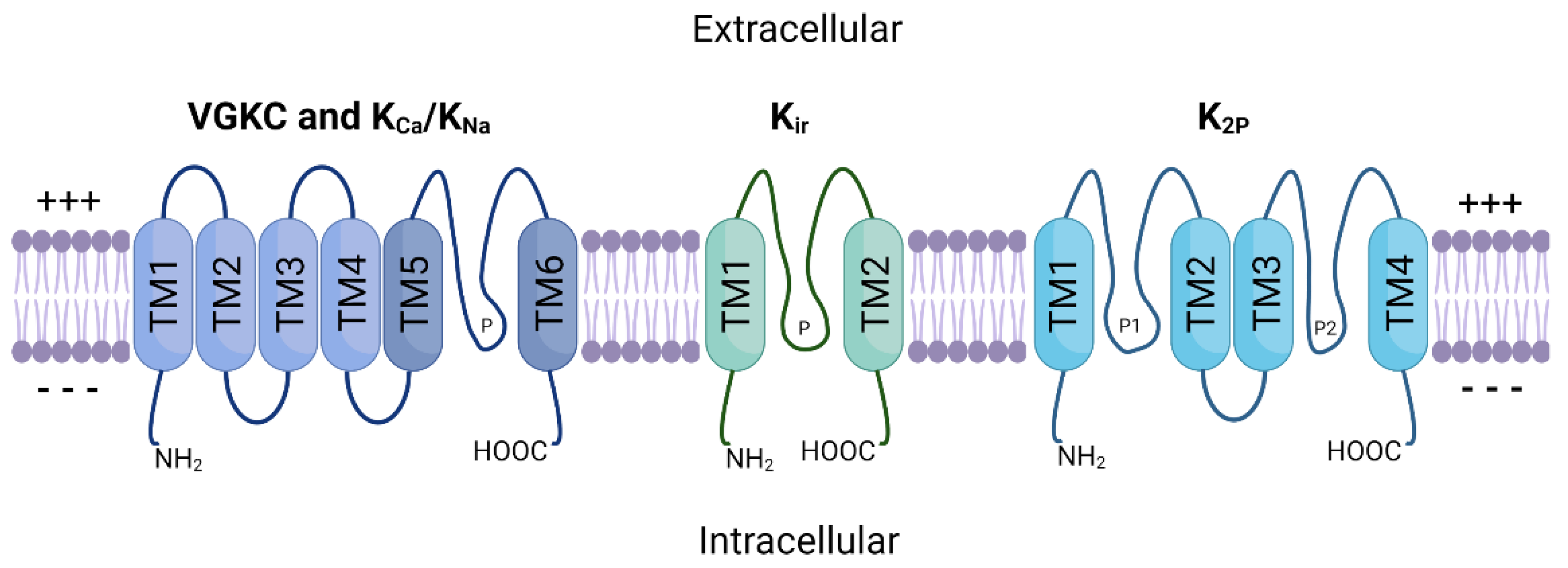
3. Malignant CNS Cancer
3.1. Targeted Therapies
3.2. Cellular Plasticity and Drug Resistance
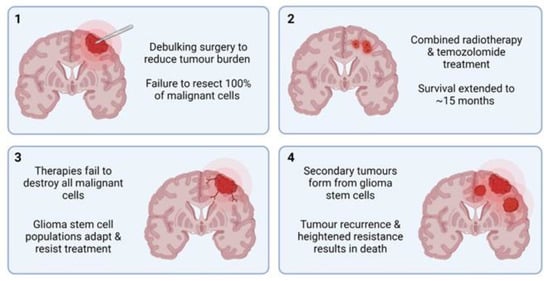
Both healthy and malignant neural cells are highly plastic, a significant barrier preventing the success of targeted HGG therapy. Sturm et al. [141][52] identified the six distinct states, differentiated by their DNA methylation patterns, between which HGG cells continually and reversibly transition. This plasticity underpins adaptive drug resistance, in which alternative signalling pathways are activated in response to the targeted blockade of integral proteins [142,143,144][53][54][55]. This process largely relies on glioma stem cells (GSCs), unique malignant cell populations capable of forming heterogenous HGG tumours. Many cancerous cells that remain following standard HGG treatment can be classified as GSCs. They are characterised by their heightened resistance to drugs, unlimited proliferation, and multipotent differentiation [145][56]. GSCs are believed to be responsible for HGG’s adaptive response to treatment by developing alternative oncogenic pathways that bypass the targeted proteins or pathways [148,149][57][58]. For example, TMZ resistance is common in GBM tumours, and GSCs are believed to be primarily responsible for this (reviewed in [150])[59]. Firstly, whole genome sequencing of matched primary and recurrent GBM tumours showed common deleterious mutations in TP53 that originated from GSC populations [151,152][60][61]. Mutations in TP53 are common in recurrent GBM, and linked to heightened invasion, migration, proliferation and GSC propagation [153][62]. It has been established that ion channels, including potassium ion channels, are key proteins in stem cells, including GSCs. Following treatment with the KCa3.1 antagonist TRAM-34, GSC cell mobility and migration was significantly reduced [155][63]. Similarly, to glioma, MB tumours are largely heterogenous and diverse, exhibiting various cell types, including stem cell-like populations (MBSCs) [158][64]. Single-cell analysis of MB tumours following vismodegib treatment revealed an increase in the population of MBSCs, which express the oncogene, SOX2 [159][65]. SOX2 plays a significant role in cancer cell stemness and is often overexpressed in the stem-cell populations of both HGG and MB [160][66].4. Potassium Ion Channels in Malignant CNS Cancer
4.1. High-Grade Glioma
The K+-regulating ion channel family is perhaps the most well understood regarding HGG pathophysiology and tumourigenesis, although several published studies have conflicting results. For example, BK channel overexpression has been identified in biopsy samples of GBM patient tumours, with the level of overexpression directly related to increasing malignancy grade [163][67]. The importance of BK channels was further solidified by the discovery of a novel splice isoform of the protein (gBK channels) that is exclusively expressed in GBM. The gBK channels appear to be directly involved in the cellular volume/size changes required for invasion of healthy brain parenchyma. Increased cytosolic Ca2+ concentration, for example via treatment with menthol, activates gBK channels and facilitates greater GBM cell migration and invasion [164][68]. While siRNA-induced knockdown of gBK channels in GBM does not change proliferation rates [165][69], various BK channel inhibitors can decrease cellular proliferation and increase tumour shrinkage in GBM cell cultures [166][70]. Conversely, BK channel activators were shown to reduce glioma cell migration by as much as 50%, suggesting that increased BK channel activity may, in fact, reduce glioma invasiveness [167][71]. Inwardly rectifying K+ (Kir) channels have also been implicated in GBM progression and tumourigenesis. Kir channels are found in abundance in normal glial cells, where they produce large, inwardly rectifying K+ currents that stabilise the membrane potential to approximately −90 mV [168][72]. However, when glial cells become malignant, the cellular membrane becomes depolarised, increasing up to −20 mV [9][8]. Further studies showed that Kir channels are expressed in GBM but are localised to the nucleus rather than the plasma membrane [169][73]. This reduces the effect that Kir channel function can have on overall cellular membrane potential. It may also contribute to epileptic seizures in many patients with GBM, as mislocalisation of Kir proteins can result in increased extracellular K+ ion concentration, which is associated with spontaneous seizures [170][74]. Thus, although Kir channels may not be as directly related to tumourigenesis as some other ion channel subtypes, they may represent potential targets in treating GBM-induced seizures.4.2. Low-Grade Glioma
Compared to HGG, there is significantly less information available regarding how potassium ion channels contribute to LGG disease progression. However, a few studies have explored how specific potassium ion channel subtypes are expressed in LGG. Diffuse grade II IDH1-mutant gliomas are particularly rare, and often affect young adults. These tumours are particularly heterogenous, displaying multiple different cell subtypes within a single tumour, but the underlying mechanisms of this were not well understood. Augustus et al. [182][75] identified that multiple proteins were involved in developing this intratumoural diversity, including KCa2.3. In the astrocyte-like tumoural cell populations, electrically active KCa2.3 channels were highly expressed, while the oligodendrocyte-like tumour cells showed significantly lower expression. This suggests that KCa2.3 may have potential to be used as a marker for astrocyte-like tumoural cells, aiding in patient tumour classification and therapeutic strategies.4.3. Medulloblastoma
The majority of existing studies targeted at potassium ion channels and medulloblastoma have centred around the Kv11.1 subtype. Huang et al. [106][76] were the first to observe that Kv11.1 was overexpressed in MB tumours samples derived from various subgroups compared with healthy brain. Kv11.1 knockdown reduced tumour cell growth in vitro and reduced tumour burden in xenograft mouse models. Kv11.1 was confined inside the cell before the G2 phase of the cell cycle, and upon reaching late G2, it was trafficked to the plasma membrane. Therefore, Kv11.1 knockdown induced G2 arrest, resulting in mitotic catastrophe and cell death. Further studies established that Kv11.1 was specifically enriched in the most invasive subpopulations of MB cells, where it regulated changes to cell shape and volume, thereby facilitating migration [186][77]. This process is facilitated by the chloride ion channel, CLIC1, during which both proteins aggregate at lipid rafts to regulate cell volume [187][78]. Simultaneous knockdown Kv11.1 and CLIC1 resulted in the suppressed growth/tumour burden in human MB cell lines and fruit fly models. Similarly, loss of the VGKC subtype Kv2.2 results in significantly improved survival in mouse models of MB [188][79].5. Potassium Ion Channels as Therapeutic Targets
Ion channels represent one of the most common targets of currently approved drugs, second only to G-coupled protein receptors [190][80]. Several potassium ion channel antagonists, categorised as class III antiarrhythmic agents, have been developed to treat cardiac arrhythmias. These include dofetilide, amiodarone, sotalol, and ibutilide, which act on the Kv11.1, 11.2, and 11.3 VGKCs, respectively, but have known off-target actions other proteins [191][81]. It is well established that Kv11 channels are responsible for mediating the heart’s inwardly rectifying K+ current. Furthermore, missense mutations in these channels can result in type 2 long QT syndrome, triggering arrhythmias [192][82]. Due to their integral role in maintaining K+ homeostasis in the heart, these drugs have potential serious adverse effects and can cause proarrhythmia [193][83]. Not all ion channel drugs block channel function. A small group of FDA-approved drugs work by forcing the channel to remain open, allowing increased K+ ion flow. Most of these drugs, including minoxidil (targets Kir1.1) and diazoxide (targets Kir6.2), are classed as vasodilators and are used to regulate blood pressure, while ezogabine targets Kv7.2 to 7.5 and acts as an anti-convulsant [191][81]. Alteration of VGKC function via blockade or sustained opening of the channel is of clinical value across several conditions. This is promising regarding CNS cancer treatment because these malignancies appear to be reliant on VGKC function for both growth and metastasis. Furthermore, because many potassium ion channel antagonists are already approved for clinical use in humans, there is potential for rapid clinical translation via drug repurposing. This includes the class III antiarrhythmics, as well as nateglinide and repaglinide, used in the management of diabetes [194][84]. When an existing drug is repurposed for a new function, such as cancer treatment, the process involved in receiving FDA approval for the new indication/s is vastly shorter than it would be for an entirely new molecule. With further research, there is potential for developing novel drugs that target specific potassium ion channel subtypes in several CNS cancers.6. Conclusions
Thanks to the collaborative efforts of numerous research teams, ourthe understanding of the roles of ion channels in tumourigenesis has grown substantially in recent decades. The discovery that ion channels act as crucial drivers of homeostasis via proliferation and cell cycle regulation, and not just as propagators of action potentials, provided the basis from which much of ourthe understanding has grown. It is now understood that ion channels are integral to the processes underlying cellular plasticity, the primary hurdle preventing the successful and curative treatment of HGG. Malignant HGG cells repurpose the endogenously expressed ion channels in healthy glial cells to further their development. Potassium ion channels are perhaps the most well-studied ion channel class in HGG oncogenesis, with many individual studies demonstrating how these cells utilise VGKC, KCa, and Kir channels for enhanced growth and metastasis. Several VGKC-targeting drugs have already received FDA approval, proving that alteration of their normal activity is possible without causing unwanted off-target effects. Further research should aim to elucidate the roles that potassium ion channels play in CNS cancers and to understand the cellular pathways and downstream proteins they influence. siRNA or CRISPR-mediated knockout studies involving key potassium ion channel subtypes in GBM cell lines would provide further insight into their involvement in key malignant cellular processes. Electrophysiological studies could also help determine whether or not these channels act independently of their current-modifying ion channel functions. Pharmacological inhibition of specific subtypes using repurposed drugs may also demonstrate the potential therapeutic value of these channels in GBM treatment. Finally, the development and use of accurate in vivo models of highly malignant tumours would be of great value, as studies involving cell lines alone can be somewhat limited in scope and potential.References
- Gabashvili, I.S.; Sokolowski, B.H.A.; Morton, C.C.; Giersch, A.B.S. Ion Channel Gene Expression in the Inner Ear. J. Assoc. Res. Otolaryngol. 2007, 8, 305–328.
- Tian, C.; Zhu, R.; Zhu, L.; Qiu, T.; Cao, Z.; Kang, T. Potassium Channels: Structures, Diseases, and Modulators. Chem. Biol. Drug Des. 2014, 83, 1–26.
- Alberts, B.; Johnson, A.; Lewis, J.; Raff, M.; Roberts, K.; Walter, P. Ion Channels and the Electrical Properties of Membranes. In Molecular Biology of the Cell, 4th ed.; Garland Science: New York, NY, USA, 2002.
- Chrysafides, S.M.; Bordes, S.; Sharma, S. Physiology, Resting Potential. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022.
- Deemyad, T.; Lüthi, J.; Spruston, N. Astrocytes Integrate and Drive Action Potential Firing in Inhibitory Subnetworks. Nat. Commun. 2018, 9, 4336.
- Grant, A.O. Cardiac Ion Channels. Circ. Arrhythm. Electrophysiol. 2009, 2, 185–194.
- Zavodnik, I.B.; Piasecka, A.; Szosland, K.; Bryszewska, M. Human Red Blood Cell Membrane Potential and Fluidity in Glucose Solutions. Scand. J. Clin. Lab. Investig. 1997, 57, 59–63.
- Bordey, A.; Sontheimer, H. Electrophysiological Properties of Human Astrocytic Tumor Cells In Situ: Enigma of Spiking Glial Cells. J. Neurophysiol. 1998, 79, 2782–2793.
- Kim, J.-B. Channelopathies. Korean J. Pediatr. 2014, 57, 1–18.
- Prevarskaya, N.; Skryma, R.; Shuba, Y. Ion Channels and the Hallmarks of Cancer. Trends Mol. Med. 2010, 16, 107–121.
- Hanahan, D.; Weinberg, R.A. The Hallmarks of Cancer. Cell 2000, 100, 57–70.
- Jackson, W.F. Potassium Channels in Regulation of Vascular Smooth Muscle Contraction and Growth. Adv. Pharmacol. 2017, 78, 89–144.
- Neylon, C.B.; Lang, R.J.; Fu, Y.; Bobik, A.; Reinhart, P.H. Molecular Cloning and Characterization of the Intermediate-Conductance Ca2+-Activated K+ Channel in Vascular Smooth Muscle: Relationship between K(Ca) Channel Diversity and Smooth Muscle Cell Function. Circ. Res. 1999, 85, e33–e43.
- Bi, D.; Toyama, K.; Lemaître, V.; Takai, J.; Fan, F.; Jenkins, D.P.; Wulff, H.; Gutterman, D.D.; Park, F.; Miura, H. The Intermediate Conductance Calcium-Activated Potassium Channel KCa3.1 Regulates Vascular Smooth Muscle Cell Proliferation via Controlling Calcium-Dependent Signaling. J. Biol. Chem. 2013, 288, 15843–15853.
- Kuang, Q.; Purhonen, P.; Hebert, H. Structure of Potassium Channels. Cell. Mol. Life Sci. 2015, 72, 3677–3693.
- Sun, X.; Zaydman, M.A.; Cui, J. Regulation of Voltage-Activated K+ Channel Gating by Transmembrane β Subunits. Front. Pharmacol. 2012, 3, 63.
- Jiang, Y.; Lee, A.; Chen, J.; Ruta, V.; Cadene, M.; Chait, B.T.; MacKinnon, R. X-ray Structure of a Voltage-Dependent K+ Channel. Nature 2003, 423, 33–41.
- Isacoff, E.Y.; Jan, L.Y.; Minor, D.L. Conduits of Life’s Spark: A Perspective on Ion Channel Research since the Birth of Neuron. Neuron 2013, 80, 658–674.
- Kaczmarek, L.K. Slack, Slick and Sodium-Activated Potassium Channels. ISRN Neurosci. 2013, 2013, 354262.
- Huang, X.; Jan, L.Y. Targeting Potassium Channels in Cancer. J. Cell Biol. 2014, 206, 151–162.
- Doyle, D.A.; Cabral, J.M.; Pfuetzner, R.A.; Kuo, A.; Gulbis, J.M.; Cohen, S.L.; Chait, B.T.; MacKinnon, R. The Structure of the Potassium Channel: Molecular Basis of K+ Conduction and Selectivity. Science 1998, 280, 69–77.
- Rasmusson, R.L.; Morales, M.J.; Wang, S.; Liu, S.; Campbell, D.L.; Brahmajothi, M.V.; Strauss, H.C. Inactivation of Voltage-Gated Cardiac K+ Channels. Circ. Res. 1998, 82, 739–750.
- Sigworth, F.J. Voltage Gating of Ion Channels. Q. Rev. Biophys. 1994, 27, 1–40.
- Sabater, V.G.; Rigby, M.; Burrone, J. Voltage-Gated Potassium Channels Ensure Action Potential Shape Fidelity in Distal Axons. J. Neurosci. 2021, 41, 5372–5385.
- Blackiston, D.J.; McLaughlin, K.A.; Levin, M. Bioelectric Controls of Cell Proliferation. Cell Cycle 2009, 8, 3519–3528.
- He, K.; McCord, M.C.; Hartnett, K.A.; Aizenman, E. Regulation of Pro-Apoptotic Phosphorylation of Kv2.1 K+ Channels. PLoS ONE 2015, 10, e0129498.
- Wang, Q.; Curran, M.E.; Splawski, I.; Burn, T.C.; Millholland, J.M.; VanRaay, T.J.; Shen, J.; Timothy, K.W.; Vincent, G.M.; de Jager, T.; et al. Positional Cloning of a Novel Potassium Channel Gene: KVLQT1 Mutations Cause Cardiac Arrhythmias. Nat. Genet. 1996, 12, 17–23.
- Kshatri, A.S.; Gonzalez-Hernandez, A.; Giraldez, T. Physiological Roles and Therapeutic Potential of Ca2+ Activated Potassium Channels in the Nervous System. Front. Mol. Neurosci. 2018, 11, 258.
- Sweet, T.-B.; Cox, D.H. Measuring the Influence of the BKCa β1 Subunit on Ca2+ Binding to the BKCa Channel. J. Gen. Physiol. 2009, 133, 139–150.
- Lee, U.S.; Cui, J. BK Channel Activation: Structural and Functional Insights. Trends Neurosci. 2010, 33, 415–423.
- Hager, N.A.; McAtee, C.K.; Lesko, M.A.; O’Donnell, A.F. Inwardly Rectifying Potassium Channel Kir2.1 and Its “Kir-Ious” Regulation by Protein Trafficking and Roles in Development and Disease. Front. Cell Dev. Biol. 2021, 9, 796136.
- Belus, M.T.; Rogers, M.A.; Elzubeir, A.; Josey, M.; Rose, S.; Andreeva, V.; Yelick, P.C.; Bates, E.A. Kir2.1 Is Important for Efficient BMP Signaling in Mammalian Face Development. Dev. Biol. 2018, 444 (Suppl. 1), S297–S307.
- Feliciangeli, S.; Chatelain, F.C.; Bichet, D.; Lesage, F. The Family of K2P Channels: Salient Structural and Functional Properties. J. Physiol. 2015, 593, 2587–2603.
- Zúñiga, L.; Zúñiga, R. Understanding the Cap Structure in K2P Channels. Front. Physiol. 2016, 7, 228.
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249.
- Miller, K.D.; Ostrom, Q.T.; Kruchko, C.; Patil, N.; Tihan, T.; Cioffi, G.; Fuchs, H.E.; Waite, K.A.; Jemal, A.; Siegel, R.L.; et al. Brain and Other Central Nervous System Tumor Statistics, 2021. CA Cancer J. Clin. 2021, 71, 381–406.
- Liu, Y.; Tyler, E.; Lustick, M.; Klein, D.; Walter, K.A. Healthcare Costs for High-Grade Glioma. Anticancer Res. 2019, 39, 1375–1381.
- Stupp, R.; Brada, M.; van den Bent, M.J.; Tonn, J.-C.; Pentheroudakis, G.; ESMO Guidelines Working Group. High-Grade Glioma: ESMO Clinical Practice Guidelines for Diagnosis, Treatment and Follow-Up. Ann. Oncol. 2014, 25 (Suppl. 3), iii93–iii101.
- Yao, M.; Li, S.; Wu, X.; Diao, S.; Zhang, G.; He, H.; Bian, L.; Lu, Y. Cellular Origin of Glioblastoma and Its Implication in Precision Therapy. Cell. Mol. Immunol. 2018, 15, 737–739.
- Wang, Y.; Jiang, T. Understanding High Grade Glioma: Molecular Mechanism, Therapy and Comprehensive Management. Cancer Lett. 2013, 331, 139–146.
- Shergalis, A.; Bankhead, A.; Luesakul, U.; Muangsin, N.; Neamati, N. Current Challenges and Opportunities in Treating Glioblastoma. Pharmacol. Rev. 2018, 70, 412–445.
- Chen, R.; Cohen, A.L.; Colman, H. Targeted Therapeutics in Patients with High-Grade Gliomas: Past, Present, and Future. Curr. Treat. Options Oncol. 2016, 17, 42.
- An, Z.; Aksoy, O.; Zheng, T.; Fan, Q.-W.; Weiss, W.A. Epidermal Growth Factor Receptor and EGFRvIII in Glioblastoma: Signaling Pathways and Targeted Therapies. Oncogene 2018, 37, 1561–1575.
- Swartz, A.M.; Li, Q.-J.; Sampson, J.H. Rindopepimut: A Promising Immunotherapeutic for the Treatment of Glioblastoma Multiforme. Immunotherapy 2014, 6, 679–690.
- Inman, S. Rindopepimut Misses OS Endpoint in Phase III Glioblastoma Trial. Available online: https://www.onclive.com/view/rindopepimut-misses-os-endpoint-in-phase-iii-glioblastoma-trial (accessed on 11 June 2022).
- Batchelor, T.T.; Mulholland, P.; Neyns, B.; Nabors, L.B.; Campone, M.; Wick, A.; Mason, W.; Mikkelsen, T.; Phuphanich, S.; Ashby, L.S.; et al. Phase III Randomized Trial Comparing the Efficacy of Cediranib as Monotherapy, and in Combination with Lomustine, versus Lomustine Alone in Patients with Recurrent Glioblastoma. J. Clin. Oncol. 2013, 31, 3212–3218.
- Gilbert, M.R.; Dignam, J.J.; Armstrong, T.S.; Wefel, J.S.; Blumenthal, D.T.; Vogelbaum, M.A.; Colman, H.; Chakravarti, A.; Pugh, S.; Won, M.; et al. A Randomized Trial of Bevacizumab for Newly Diagnosed Glioblastoma. N. Engl. J. Med. 2014, 370, 699–708.
- Chinot, O.L.; Wick, W.; Mason, W.; Henriksson, R.; Saran, F.; Nishikawa, R.; Carpentier, A.F.; Hoang-Xuan, K.; Kavan, P.; Cernea, D.; et al. Bevacizumab plus Radiotherapy-Temozolomide for Newly Diagnosed Glioblastoma. N. Engl. J. Med. 2014, 370, 709–722.
- Neftel, C.; Laffy, J.; Filbin, M.G.; Hara, T.; Shore, M.E.; Rahme, G.J.; Richman, A.R.; Silverbush, D.; Shaw, M.L.; Hebert, C.M.; et al. An Integrative Model of Cellular States, Plasticity, and Genetics for Glioblastoma. Cell 2019, 178, 835–849.
- Debanne, D.; Daoudal, G.; Sourdet, V.; Russier, M. Brain Plasticity and Ion Channels. J. Physiol. Paris 2003, 97, 403–414.
- Liebau, S.; Kleger, A.; Levin, M.; Yu, S.P. Stem Cells and Ion Channels. Stem Cells Int. 2013, 2013, 238635.
- Sturm, D.; Witt, H.; Hovestadt, V.; Khuong-Quang, D.-A.; Jones, D.T.W.; Konermann, C.; Pfaff, E.; Tönjes, M.; Sill, M.; Bender, S.; et al. Hotspot Mutations in H3F3A and IDH1 Define Distinct Epigenetic and Biological Subgroups of Glioblastoma. Cancer Cell 2012, 22, 425–437.
- Greenall, S.A.; Donoghue, J.F.; Van Sinderen, M.; Dubljevic, V.; Budiman, S.; Devlin, M.; Street, I.; Adams, T.E.; Johns, T.G. EGFRvIII-Mediated Transactivation of Receptor Tyrosine Kinases in Glioma: Mechanism and Therapeutic Implications. Oncogene 2015, 34, 5277–5287.
- Pillay, V.; Allaf, L.; Wilding, A.L.; Donoghue, J.F.; Court, N.W.; Greenall, S.A.; Scott, A.M.; Johns, T.G. The Plasticity of Oncogene Addiction: Implications for Targeted Therapies Directed to Receptor Tyrosine Kinases. Neoplasia 2009, 11, 448–458.
- Friedmann-Morvinski, D. Glioblastoma Heterogeneity and Cancer Cell Plasticity. Crit. Rev. Oncog. 2014, 19, 327–336.
- Osuka, S.; Meir, E.G.V. Overcoming Therapeutic Resistance in Glioblastoma: The Way Forward. J. Clin. Investig. 2017, 127, 415–426.
- Lathia, J.D.; Mack, S.C.; Mulkearns-Hubert, E.E.; Valentim, C.L.L.; Rich, J.N. Cancer Stem Cells in Glioblastoma. Genes Dev. 2015, 29, 1203–1217.
- Hadjipanayis, C.G.; Van Meir, E.G. Tumor Initiating Cells in Malignant Gliomas: Biology and Implications for Therapy. J. Mol. Med. 2009, 87, 363–374.
- Singh, N.; Miner, A.; Hennis, L.; Mittal, S. Mechanisms of Temozolomide Resistance in Glioblastoma—A Comprehensive Review. Cancer Drug Resist. 2021, 4, 17–43.
- Kim, H.; Zheng, S.; Amini, S.S.; Virk, S.M.; Mikkelsen, T.; Brat, D.J.; Grimsby, J.; Sougnez, C.; Muller, F.; Hu, J.; et al. Whole-Genome and Multisector Exome Sequencing of Primary and Post-Treatment Glioblastoma Reveals Patterns of Tumor Evolution. Genome Res. 2015, 25, 316–327.
- Orzan, F.; De Bacco, F.; Crisafulli, G.; Pellegatta, S.; Mussolin, B.; Siravegna, G.; D’Ambrosio, A.; Comoglio, P.M.; Finocchiaro, G.; Boccaccio, C. Genetic Evolution of Glioblastoma Stem-Like Cells from Primary to Recurrent Tumor. Stem Cells 2017, 35, 2218–2228.
- Zhang, Y.; Dube, C.; Gibert, M.; Cruickshanks, N.; Wang, B.; Coughlan, M.; Yang, Y.; Setiady, I.; Deveau, C.; Saoud, K.; et al. The P53 Pathway in Glioblastoma. Cancers 2018, 10, E297.
- Ruggieri, P.; Mangino, G.; Fioretti, B.; Catacuzzeno, L.; Puca, R.; Ponti, D.; Miscusi, M.; Franciolini, F.; Ragona, G.; Calogero, A. The Inhibition of KCa3.1 Channels Activity Reduces Cell Motility in Glioblastoma Derived Cancer Stem Cells. PLoS ONE 2012, 7, e47825.
- Ocasio, J.K.; Babcock, B.; Malawsky, D.; Weir, S.J.; Loo, L.; Simon, J.M.; Zylka, M.J.; Hwang, D.; Dismuke, T.; Sokolsky, M.; et al. ScRNA-Seq in Medulloblastoma Shows Cellular Heterogeneity and Lineage Expansion Support Resistance to SHH Inhibitor Therapy. Nat. Commun. 2019, 10, 5829.
- Vanner, R.J.; Remke, M.; Gallo, M.; Selvadurai, H.J.; Coutinho, F.; Lee, L.; Kushida, M.; Head, R.; Morrissy, S.; Zhu, X.; et al. Quiescent Sox2+ Cells Drive Hierarchical Growth and Relapse in Sonic Hedgehog Subgroup Medulloblastoma. Cancer Cell 2014, 26, 33–47.
- Mansouri, S.; Nejad, R.; Karabork, M.; Ekinci, C.; Solaroglu, I.; Aldape, K.D.; Zadeh, G. Sox2: Regulation of Expression and Contribution to Brain Tumors. CNS Oncol. 2016, 5, 159–173.
- Ransom, C.B.; Sontheimer, H. BK Channels in Human Glioma Cells. J. Neurophysiol. 2001, 85, 790–803.
- Wondergem, R.; Bartley, J.W. Menthol Increases Human Glioblastoma Intracellular Ca2+, BK Channel Activity and Cell Migration. J. Biomed. Sci. 2009, 16, 90.
- Abdullaev, I.F.; Rudkouskaya, A.; Mongin, A.A.; Kuo, Y.-H. Calcium-Activated Potassium Channels BK and IK1 Are Functionally Expressed in Human Gliomas but Do Not Regulate Cell Proliferation. PLoS ONE 2010, 5, e12304.
- Weaver, A.K.; Liu, X.; Sontheimer, H. Role for Calcium-Activated Potassium Channels (BK) in Growth Control of Human Malignant Glioma Cells. J. Neurosci. Res. 2004, 78, 224–234.
- Kraft, R.; Krause, P.; Jung, S.; Basrai, D.; Liebmann, L.; Bolz, J.; Patt, S. BK Channel Openers Inhibit Migration of Human Glioma Cells. Pflug. Arch. 2003, 446, 248–255.
- Newman, E.A. Inward-Rectifying Potassium Channels in Retinal Glial (Müller) Cells. J. Neurosci. 1993, 13, 3333–3345.
- Olsen, M.L.; Sontheimer, H. Mislocalization of Kir Channels in Malignant Glia. Glia 2004, 46, 63–73.
- Traynelis, S.F.; Dingledine, R. Potassium-Induced Spontaneous Electrographic Seizures in the Rat Hippocampal Slice. J. Neurophysiol. 1988, 59, 259–276.
- Augustus, M.; Pineau, D.; Aimond, F.; Azar, S.; Lecca, D.; Scamps, F.; Muxel, S.; Darlix, A.; Ritchie, W.; Gozé, C.; et al. Identification of CRYAB+ KCNN3+ SOX9+ Astrocyte-Like and EGFR+ PDGFRA+ OLIG1+ Oligodendrocyte-Like Tumoral Cells in Diffuse IDH1-Mutant Gliomas and Implication of NOTCH1 Signalling in Their Genesis. Cancers 2021, 13, 2107.
- Huang, X.; Dubuc, A.M.; Hashizume, R.; Berg, J.; He, Y.; Wang, J.; Chiang, C.; Cooper, M.K.; Northcott, P.A.; Taylor, M.D.; et al. Voltage-Gated Potassium Channel EAG2 Controls Mitotic Entry and Tumor Growth in Medulloblastoma via Regulating Cell Volume Dynamics. Genes Dev. 2012, 26, 1780–1796.
- Huang, X.; He, Y.; Dubuc, A.M.; Hashizume, R.; Zhang, W.; Reimand, J.; Yang, H.; Wang, T.A.; Stehbens, S.J.; Younger, S.; et al. EAG2 Potassium Channel with Evolutionarily Conserved Function as a Brain Tumor Target. Nat. Neurosci. 2015, 18, 1236–1246.
- Francisco, M.A.; Wanggou, S.; Fan, J.J.; Dong, W.; Chen, X.; Momin, A.; Abeysundara, N.; Min, H.-K.; Chan, J.; McAdam, R.; et al. Chloride Intracellular Channel 1 Cooperates with Potassium Channel EAG2 to Promote Medulloblastoma Growth. J. Exp. Med. 2020, 217, e20190971.
- Fan, J.; Pusong, R.; Morrissy, S.; Farooq, H.; Taylor, M.; Huang, X. MEDU-28. Eliminating the Root of Medulloblastoma by Targeting a Voltage-Gated Potassium Channel. Neuro Oncol. 2019, 21, ii109.
- Santos, R.; Ursu, O.; Gaulton, A.; Bento, A.P.; Donadi, R.S.; Bologa, C.G.; Karlsson, A.; Al-Lazikani, B.; Hersey, A.; Oprea, T.I.; et al. A Comprehensive Map of Molecular Drug Targets. Nat. Rev. Drug Discov. 2017, 16, 19–34.
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A Major Update to the DrugBank Database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082.
- Vandenberg, J.I.; Perry, M.D.; Perrin, M.J.; Mann, S.A.; Ke, Y.; Hill, A.P. HERG K+ Channels: Structure, Function, and Clinical Significance. Physiol. Rev. 2012, 92, 1393–1478.
- Brendorp, B.; Pedersen, O.; Torp-Pedersen, C.; Sahebzadah, N.; Køber, L. A Benefit-Risk Assessment of Class III Antiarrhythmic Agents. Drug Saf. 2002, 25, 847–865.
- Guardado-Mendoza, R.; Prioletta, A.; Jiménez-Ceja, L.M.; Sosale, A.; Folli, F. The Role of Nateglinide and Repaglinide, Derivatives of Meglitinide, in the Treatment of Type 2 Diabetes Mellitus. Arch. Med. Sci. 2013, 9, 936–943.