Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Lingling Ge | -- | 2958 | 2022-09-24 12:03:27 | | | |
| 2 | Jessie Wu | + 8 word(s) | 2966 | 2022-09-26 07:13:58 | | | | |
| 3 | Jessie Wu | + 2 word(s) | 2968 | 2022-09-26 07:17:23 | | | | |
| 4 | Jessie Wu | Meta information modification | 2968 | 2022-09-26 07:25:36 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Ge, L.; Xing, M.; Zhang, H.; Wang, Z. Neurofibroma Development in Neurofibromatosis Type 1. Encyclopedia. Available online: https://encyclopedia.pub/entry/27553 (accessed on 25 July 2026).
Ge L, Xing M, Zhang H, Wang Z. Neurofibroma Development in Neurofibromatosis Type 1. Encyclopedia. Available at: https://encyclopedia.pub/entry/27553. Accessed July 25, 2026.
Ge, Ling-Ling, Ming-Yan Xing, Hai-Bing Zhang, Zhi-Chao Wang. "Neurofibroma Development in Neurofibromatosis Type 1" Encyclopedia, https://encyclopedia.pub/entry/27553 (accessed July 25, 2026).
Ge, L., Xing, M., Zhang, H., & Wang, Z. (2022, September 24). Neurofibroma Development in Neurofibromatosis Type 1. In Encyclopedia. https://encyclopedia.pub/entry/27553
Ge, Ling-Ling, et al. "Neurofibroma Development in Neurofibromatosis Type 1." Encyclopedia. Web. 24 September, 2022.
Copy Citation
Neurofibromatosis type 1 (NF1), a genetic tumor predisposition syndrome that affects about 1 in 3000 newborns, is caused by mutations in the NF1 gene and subsequent inactivation of its encoded neurofibromin. Neurofibromin is a tumor suppressor protein involved in the downregulation of Ras signaling. Despite a diverse clinical spectrum, one of several hallmarks of NF1 is a peripheral nerve sheath tumor (PNST), which comprises mixed nervous and fibrous components. The distinct spatiotemporal characteristics of plexiform and cutaneous neurofibromas have prompted hypotheses about the origin and developmental features of these tumors, involving various cellular transition processes.
NF1
neurofibroma formation
SCPs
1. Neurofibroma Formation
1.1. The Developmental Origin of Schwann Cell Lineages
Friederich von Recklinghausen initially coined the concept of neurofibroma in 1882 [1], noting that both neuronal and fibrotic components were present within these tumors. In subsequent studies, the identification of abnormal Schwann cell (SC) proliferation in neurofibromas led to the SC origin hypothesis [2]; therefore, neurofibromas have long been recognized to originate from SC lineages. Despite the early consideration of mature SCs as the pathogenic origin, studies published recently following the establishment of various genetically engineered mouse (GEM) models indicate the possibility that neurofibromas may originate from earlier-stage SCs [3][4][5][6][7]. To date, the specific cell type within the SC lineage leading to neurofibroma formation is controversial.
The term neural crest stem cell (NCSC) was first put forward by Stemple and Anderson in 1992, following their successful isolation of neural crest cell populations with self-renewal ability and multipotency in vitro [8]. NCSCs are a transient cell population, emerging at the dorsolateral portion of the neural tube during vertebrate embryogenesis and then migrating to extensive locations. They later differentiate into a wide range of cell lineages and tissues, depending on the local environment, including most of the neuronal and glial components of the peripheral nervous system (PNS), as well as bone, cartilage, endocrine cells, melanocytes, fibroblasts, and smooth muscle cells [9][10][11].
In the first stage of SC lineage development, a subpopulation of NCSCs gives rise to boundary cap (BC) cells. These are transiently located at the motor exit point (MEP) and the dorsal root entry zone (DREZ), acting as a boundary between the central and peripheral nervous systems and allowing the passage of axons [12][13]. The discovery of specific molecular markers has greatly contributed to the further characterization of BC cells [14]. These cells express the transcription factor gene Krox20, also known as EGR2 in humans, and produce the SC components of the dorsal and ventral nerve roots, playing a role in the early myelination of the PNS [15]. Moreover, in culture, BC cells can also generate other cell types, such as melanocytes, astrocytes, and neurons [14][16]. In addition, a subpopulation of BC derivatives was recently found to express Prss56; lineage-tracing studies demonstrated that Prss56-expressing BCs have broad differentiation potential and can give rise to SCs in the nerve roots, hypodermis, and dermis, suggesting the potential of BC cells as candidates for the cellular origin of both pNFs and cNFs [17]. The specific expression pattern of Krox20/EGR2 and Prss56, together with Hey2 and Wif1 in mouse and/or human lines, suggests that BC clusters emerge at embryonic day (E) 10.5–11 in mice [18].
In addition to differentiation into BC cells, migrating NCSCs (both multipotent and restricted) can differentiate into Schwann cell precursors (SCPs) at around E12 to E13 in mice [19]. Furthermore, both Krox20-expressing and Prss56-expressing BC cells can convert to SCPs in nerve roots and to satellite cells and nociceptive neurons in the dorsal root ganglia (DRG) [14][20][21]. SCPs are glial-restricted cells found in early embryonic nerves, which are in intimate contact with nerve axons and maintain a certain level of multipotency; they have the ability to generate endoneurial fibroblasts, melanocytes, and parasympathetic or enteric neurons. Although they share some common features with NCSCs, SCPs differ in the expression of specific glial differentiation genes and molecular markers, such as myelin protein 0 (P0), growth-associated protein 43 (GAP43), cadherin-19, and other molecular factors [22]. Another specific characteristic of SCPs is their dependence on axon-associated signals, which determine their proliferation and differentiation to myelinating or non-myelinating cells [23]. In the second stage of SC lineage development, a subset of SCPs converts into immature SCs at E13–15 in mice, regulated by a number of signals associated with axons, including neuregulin 1 (NRG1), endothelin, and the notch signaling pathway. Similar to SCPs, immature SCs maintain close contact with axons but differ substantially in their molecular phenotype, with increased expression of specific proteins, including glial fibrillary acidic protein (GFAP) and S100 calcium-binding protein (S100). In addition, the survival of immature SCs depends on autocrine signals, rather than axon-associated NRG1 signals.
In the subsequent stage, the associated axons determine the developmental type of immature SCs [24]. Immature SCs that are in contact with large-diameter axons, reaching a ratio of 1:1 through proliferation, and proceed to transform into myelinating SCs (mSCs) around birth [25]. In contrast, immature SCs in contact with small-diameter axons develop into mature non-myelinating SCs (nmSCs) at varying SC-to-axon ratios and form Remak bundles [23] (Figure 1).
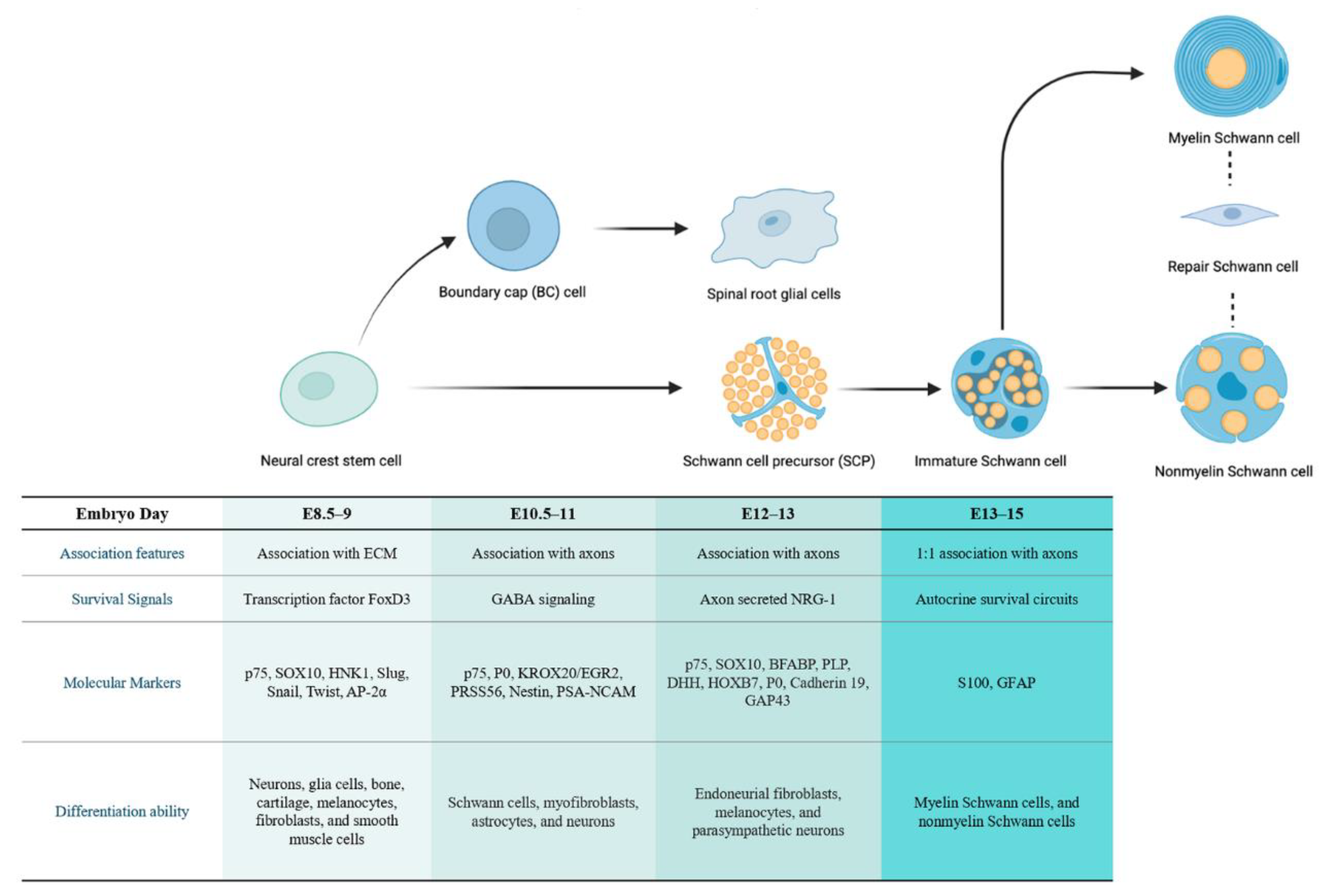
Figure 1. The developmental stage of SC lineage and corresponding characteristics of different cell types. Neural crest stem cells (NCSCs) can differentiate into multipotent boundary cap (BC) cells and SC precursors (SCPs). The SCPs further develop into immature SCs, which then differentiate into myelinating/non-myelinating SCs according to the associated axons. These mature types can de-differentiate upon specific mutation or injury into repair SCs. The corresponding embryogenesis time of each cell type in mice and other features, including their association characteristics, survival signals, molecular markers, and differentiation capacity, are listed relative to the cells.
1.1.1. The Cellular Origin of Neurofibroma
The cutaneous form of NF occurs in almost all NF1 patients, with tumors typically emerging around puberty and potentially increasing in number over the lifespan of the patients. In contrast, pNFs arise in around 30% of NF1 patients from early childhood and gradually expand throughout life. The significant differences between these two subtypes of neurofibromas and the phenomenon that mouse models develop pNF but fail to develop cNF at 100% frequency jointly indicate that the cellular origins of these lesions may differ. Specifically, their temporally and spatially distinct clinical characteristics support the hypothesis that pNFs are congenital lesions arising from the embryonic SC lineage, whereas cNFs likely derive from a more mature cell type in the SC lineage [26].
1.1.2. The Cellular Origin of pNF
Although the hypothesis of the SC origin of neurofibroma has been put forward by researchers for decades, it was not until 2002 that GEM models successfully recapitulated human pNF lesions, definitively demonstrating the potential of SCs to be the lineage of origin. Knowing the crucial role of Krox20 in SC development, Zhu and coworkers used Krox20-Cre in mouse models to specifically delete Nf1 in SC lineage cells [27]. They found that loss of Nf1 from the SC lineage in an Nf1+/− environment successfully recapitulated pNF formation in spinal nerve roots. However, although Krox20-Cre could induce pNFs, the extensive expression of Krox20 in NCSCs, SCPs, and SCs meant that the exact time of initiation and cells of origin remained unknown [15]. In 2008, Joseph et al. showed that germline deletion or conditional deletion of Nf1 using Wnt1-Cre led to transient hyperproliferation and self-renewal of NCSCs without typical tumor formation. In addition, no NCSCs were identified in normal adult peripheral nerves or the regions that develop neurofibroma, and no tumorigenicity due to Nf1 loss in NCSCs was observed. Accordingly, the authors speculated that neurofibromas might arise from later NCSC derivatives [28]. In the same year, Zheng et al. induced mutation of Nf1 in SCPs using P0a-Cre rather than the Krox20-Cre, which led to pNF formation in the sciatic nerve. The results suggested that nmSCs of the Remak bundles might be the cellular origin for neurofibroma [29]. However, no conclusion could be drawn as to which stage in the SC lineage was critical for neurofibroma formation mediated by NF1 loss. In 2011, Le and colleagues reported that inducible Plp-CreERT2-mediated ablation of Nf1 in SCs during both embryonic and adult stages resulted in peripheral nerve hyperplasia and pNF formation. However, embryonic stages (including SCPs and immature SCs) were more susceptible to pNF, in comparison with adult stages (100% versus 2%) [3]. Another study, carried out by Mayes and coworkers, proposed that embryonic and adult SCs had similar potential to give rise to neurofibromas; however, the clinical manifestation of pNFs as congenital lesions is less supportive of a central role for mature SCs [4]. In 2014, Chen et al. reported that the cells of origin for paraspinal pNF were PLP+GAP43+ cells, which could be detected in the embryonic DRG at E11.5 but not at E13.5. It was also demonstrated that PLP+ cell populations included both embryonic Krox20+ and Dhh+ cells [6]. Due to their specific expression of molecular markers, PLP+GAP43+ cells were considered to be at the SCP developmental stage and therefore potentially the elusive cells of origin for paraspinal pNF. The authors hypothesized that there may be an overlapping of cell types in the transition from NCSCs to embryonic and mature SCs, such that a subpopulation of the remaining SCPs could continue into adulthood and retain the potential for pNF formation [6].
1.1.3. The Cellular Origin of cNF
Unlike the considerable achievements made in developing GEM models to study the cellular origin of pNF, few animal models have been established to recapitulate the characteristics of cNF, leaving its origin and pathogenic mechanisms relatively unknown. Given the near 100% incidence of cNF in NF1 individuals, there remains an urgent need to investigate the formation and development of cNF. The first GEM model to successfully generate cNF was produced by Satio et al. in 2007, using Camk2-Cre to drive N-Ras activation [30]. These transgenic mice exhibited hyperpigmentation of the epidermis throughout their lives and developed diffuse cNF later on. Nonetheless, pNF lesions and other manifestations, such as schwannomas and astrocytomas, were not detected in this research. The authors speculated that further signals in addition to activated N-Ras may be required for the development of these tumors. In 2008, Wu and colleagues established a GEM model using Dhh-Cre to inactivate the Nf1 gene [31]. In vivo ablation of Nf1 at E12.5 not only recapitulated human pNF but also effectively generated cNF in an Nf1+/− microenvironment. The results obtained in these studies overturned the previous view that cNF probably arose from mature cell types in the SC lineage, based on its time of initiation [26]. Regarding the location of cNF, follow-up studies further explored its specific origin, focusing on another stem cell population known as skin-derived precursors (SKPs), found in the dermis of humans and mice. SKPs are also multipotent, with the capacity to differentiate along neuronal and glial cell lineages, giving rise to SCs, neurons, adipocytes, and other cell types. In 2009, Le et al. pioneered research into the ability of SKPs to induce neurofibromas upon Nf1 loss. In this research, SKPs isolated from tamoxifen-treated Nf1−/− CMV-CreERT2 mice that had been injected in the proximity of the sciatic nerves recapitulated pNF, indicating the intrinsic capacity of SKPs to generate neurofibromas. However, SKPs implanted in the dermis of mice could also generate classic cNF lesions [32]. These data suggested that a specific cell type within the SKP population was the cellular origin for cNF tumor initiation. However, since SKPs are a heterogeneous cell population, the essential questions of which subsets of cells give rise to which subtype of neurofibromas or whether there is a common origin within SKPs to form both cNFs and pNFs in the absence of NF1 remain to be answered.
1.1.4. Associate pNF and cNF with a Common Stage of Origin
With the discovery of SKPs as a possible common origin for the different subtypes of neurofibromas, the previous concept of distinctive initiation stages was transformed to that of a shared initiation stage. The explanation for the difference in the timing and location of occurrence was the spatiotemporal difference in NF1 loss at subsequent developmental stages. In 2019, Chen and coworkers [33], as well as Radomska and colleagues, proposed that cNFs and pNFs may originate separately from the same cell population, HoxB7/Prss56-expressing BC cells or SCPs [34]. However, despite the effective generation of pNFs and cNFs in GEM models and breakthroughs in the hypothesis of cellular origin, the distinctive phenotypes observed in mouse models and human neurofibroma require further investigation. In 2021, Mo et al. used human induced pluripotent stem cells (hiPSCs) to identify the common cells susceptible to mutation in different types of neurofibromas [35]. The results suggested that biallelic inactivation of Nf1 in SOX10+ cells of the SC lineage could lead to the formation of both cNFs and pNFs. Future investigations utilizing these hiPSC lines will allow the mechanisms that define neurofibroma formation to be better understood by applying the insights gained from studies into cellular origin.
1.2. Alterations in Schwann Cells in the Early Stage of Tumorigenesis
Under normal circumstances, SCs cover most of the surface of peripheral nerve axons, and their behavior is recognized to be adhesively controlled by axonal contact. Signals regulating survival, proliferation, and differentiation transmitted via axons during embryonic and adult stages are regarded as vital to maintaining SCs in a differentiated state and ensuring normal neural functions [24][36]. In recent years, the molecular mechanisms of SC–axonal interactions, including the NRG1-ErbB signaling pathway, have been widely studied.
Loss of contact between transiently proliferating SCs and axons is a common occurrence in the early stages of neurofibroma development [31]. A mechanistic explanation provided for this crucial event is that disruption to SC–axonal interactions results from the Ras-Raf-ERK-dependent downregulation of an SC surface protein named semaphorin 4F (Sema4F) [37]. High levels of Ras signaling and low levels of Sema4F trigger tumorigenic properties in neoplastic SCs, inducing increased proliferation. In addition to the molecular mechanisms of pNF, Radomska et al. provided a perspective on the occurrence of cNF [34]. They hypothesized that the increase in density of local innervation in mutant skin might be a mechanism to compensate for SC hyperplasia in order to maintain appropriate levels of contact; however, when overridden, SCs can no longer interact with axons, and the increased branching may lead to a pro-tumorigenic phenotype. The branching capacity of nerve terminals in the upper dermis may be associated with the lack of perineurium [34].
2. Neurofibroma Progression
2.1. Schwann Cells Contribution and Lineage Shift
In the process of neurofibroma growth and progression, SCs, the most abundant glial cells in the PNS and also the suspectable tumor cells of neurofibromas, have been shown to play multiple roles. Stonecypher et al. found that neoplastic SCs could produce NRG1, which then promoted neoplastic SC proliferation in an autocrine or paracrine way [38]. Neoplastic SCs also secreted cytokines, such as stem cell factor (SCF) and colony-stimulating factor 1 (CSF1); such factors were proposed to act in a “cytokine-cytokine receptor” manner, recruiting immune cells such as mast cells and macrophages, both of which secrete transforming growth factor-β (TGF-β) to active neurofibroma-associated fibroblasts for ECM remodeling [38]. As in the process of neurofibroma formation, a process of rapid de-differentiation of SCs is triggered by axonal damage, which subsequently destroys the myelin sheath. With the development and progression of the tumor, these SCs undergo consistent de-differentiation and finally revert to a progenitor-like state of proliferation [39]. In this process of cellular transition, the synergistic effects of the Ras-Raf-MEK-ERK pathway and inflammatory signals have been demonstrated as the driving factors [40]. Several studies have been explored to identify related inflammatory signals and determine altered gene expression patterns involved in this conversion process, including downregulation of genes coding for the key myelin transcription factor Krox20, as well as structural proteins such as P0, and upregulation of pro-inflammatory factors, such as tumor necrosis factor α (TNFα), interleukin-1α (IL-1α), and interleukin-1β (IL-1β) [34][41][42].
Specifically, additional effects of nerve injury in facilitating SC phenotype transition have also been recognized. To verify this, the researchers obtained pigmented melanocytes (probably by SC trans-differentiation) and rare neurofibroma formation after cutting the sciatic nerve in Nf1 heterozygous mice [43]. Ribeiro et al. performed nerve crush in P0-Nf1fl/fl and P0-Nf1fl/− mice that do not develop neurofibromas, and observed infiltration of immune cells and appearance of neurofibromas [44].
Wound repair following local trauma is regarded as a dynamic process followed by three main phases—inflammation, proliferation, and remodeling—in which various candidate mediators participate [45]. Thus, upon local trauma, the demand for new undifferentiated cells is met by the nerve regeneration capacity, which can promote the transformation of mSCs and Remak bundles into repair SCs, which is a pro-tumorigenic phenotype and capable of accelerating neurofibroma progression [46] (Figure 2).
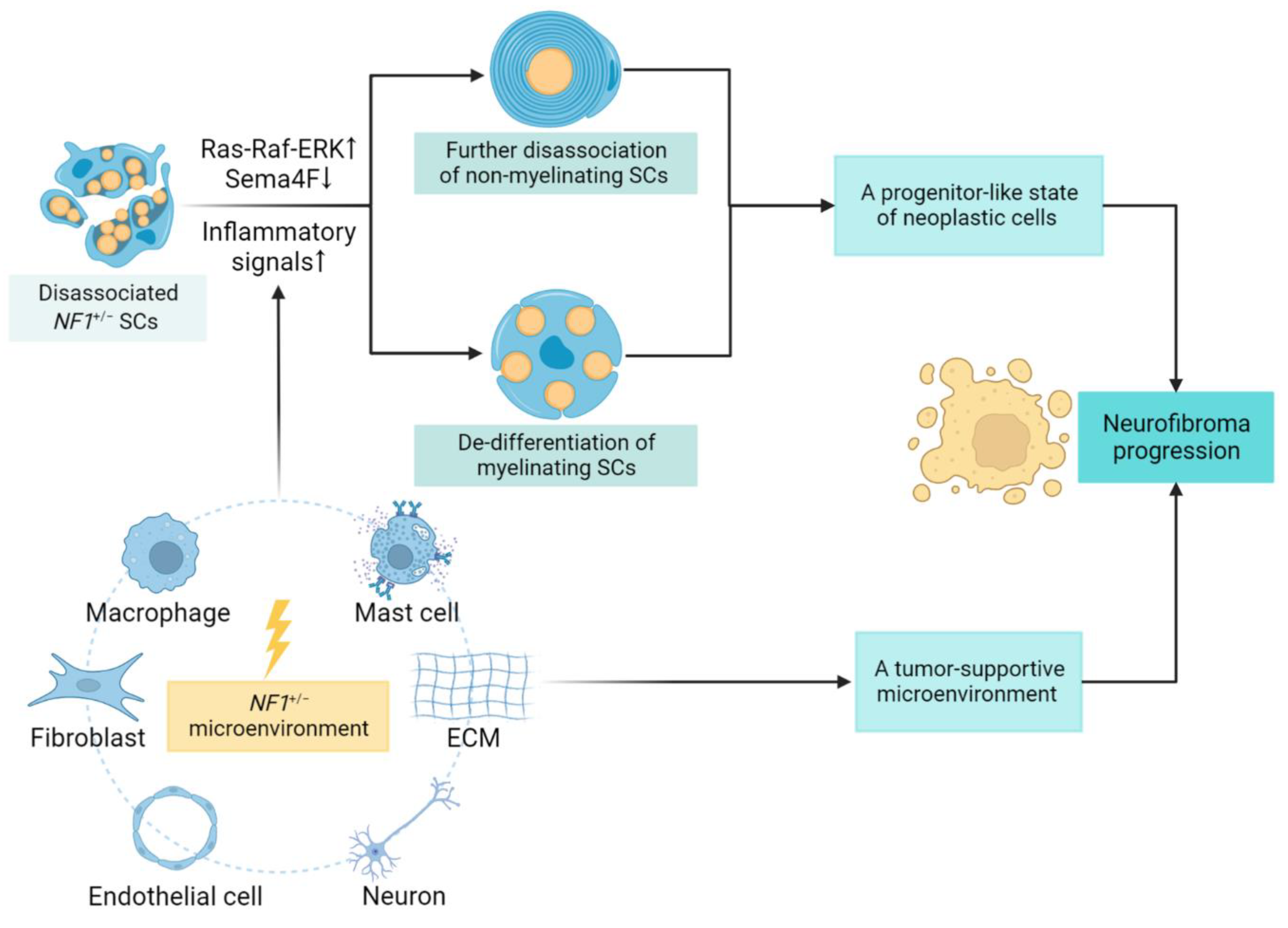
Figure 2. SC lineage shift and contributing factors in neurofibroma progression. The neoplastic SCs can rapidly de-differentiate to a progenitor-like state, disrupting SC–axonal interactions with tumor development. The underlying mechanism involves Ras-dependent downregulation of an SC surface protein, semaphorin 4F (Sema4F), together with elevated inflammatory signals, especially upon injury. Other environmental factors, including cellular and non-cellular components, further create a tumor-promoting microenvironment. The proliferative state of neoplastic cells and supportive tumor microenvironment combined to promote neurofibroma progression. ↑: upregulation of signaling pathways; ↓: downregulation in expression.
2.2. Role of the Tumor Microenvironment
During the early embryonic stages, the microenvironment appears to be tumor-suppressive, allowing normal differentiation and proliferation of NF−/− SCPs [47]. However, as neurofibromas develop, the nerve microenvironment converts to a tumor-promoting type, with complex mutual interactions between cellular and non-cellular components. As heterogeneous tumors, neurofibromas comprise neoplastic SCs as well as fibroblasts, immune cells, neurons, endothelial cells, and ECM components. In addition to the original neoplastic cells, the non-neoplastic cell types in the tumor microenvironment are also crucial in the development of neurofibromas. A series of genetic studies have demonstrated that NF1-homozygous SC lineage cells and haploinsufficiency of NF1 in non-neuronal cells are both required to promote the pathogenesis of neurofibroma [31][32][48][49][50][51]. The complex effects of the tumor microenvironment on neurofibroma formation and progression, especially the intricate interactions of both cellular and non-cellular components, have been summarized in detail in a review published in 2021 [52]; however, specific mechanisms remain unclear. Moreover, the occurrence of neurofibroma in normal individuals, as well as the recognition of patient subgroups with mosaic NF1 caused by postzygotic NF1 mutation, suggest that an NF1+/− environment may not necessarily be required for neurofibroma formation [31]. Thus, further studies and animal models are still urgently required to recapitulate the characteristics of the human neurofibroma microenvironment and shed light on its function in neurofibroma growth and progression.
References
- Crump, T.; Translation of case reports in Ueber die multiplen Fibrome der Haut und ihre Beziehung zu den multiplen Neuromen by F. v. Recklinghausen.. Advances in Neurology 1981, 29, 259-275.
- Yoshimasa Kamata; Study on the ultrastructure and acetylcholinesterase activity in von Recklinghausen's neurofibromatosis. Pathology International 1978, 28, 393-410, 10.1111/j.1440-1827.1978.tb01264.x.
- Lu Q. Le; Chiachi Liu; Tracey Shipman; Zhiguo Chen; Ueli Suter; Luis F. Parada; Susceptible Stages in Schwann Cells for NF1-Associated Plexiform Neurofibroma Development. Cancer Research 2011, 71, 4686-4695, 10.1158/0008-5472.can-10-4577.
- Debra A. Mayes; Tilat A. Rizvi; Jose A. Cancelas; Nathan T. Kolasinski; Georgianne M. Ciraolo; Anat O. Stemmer-Rachamimov; Nancy Ratner; Perinatal or adult Nf1 inactivation using tamoxifen-inducible PlpCre each cause neurofibroma formation. Cancer Research 2011, 71, 4675-4685, 10.1158/0008-5472.can-10-4558.
- Vincent W. Keng; Eric P. Rahrmann; Adrienne L. Watson; Barbara R. Tschida; Christopher L. Moertel; Walter J. Jessen; Tilat A. Rizvi; Margaret H. Collins; Nancy Ratner; David A. Largaespada; et al. PTEN and NF1 inactivation in Schwann cells produces a severe phenotype in the peripheral nervous system that promotes the development and malignant progression of peripheral nerve sheath tumors. Cancer Research 2012, 72, 3405-3413, 10.1158/0008-5472.can-11-4092.
- Zhiguo Chen; Chiachi Liu; Amish Patel; Chung-Ping Liao; Yong Wang; Lu Q. Le; Cells of Origin in the Embryonic Nerve Roots for NF1-Associated Plexiform Neurofibroma. Cancer Cell 2014, 26, 695-706, 10.1016/j.ccell.2014.09.009.
- Katherine E. Chaney; Melissa R. Perrino; Leah J. Kershner; Ami V. Patel; Jianqiang Wu; Kwangmin Choi; Tilat A. Rizvi; Eva Dombi; Sara Szabo; David A. Largaespada; et al.Nancy Ratner Cdkn2a Loss in a Model of Neurofibroma Demonstrates Stepwise Tumor Progression to Atypical Neurofibroma and MPNST. Cancer Research 2020, 80, 4720-4730, 10.1158/0008-5472.can-19-1429.
- Derek L. Stemple; David J. Anderson; Isolation of a stem cell for neurons and glia from the mammalian neural crest. Cell 1992, 71, 973-985, 10.1016/0092-8674(92)90393-q.
- Annita Achilleos; Paul A Trainor; Neural crest stem cells: discovery, properties and potential for therapy. Cell Research 2012, 22, 288-304, 10.1038/cr.2012.11.
- Ruslan Soldatov; Marketa Kaucka; Maria Eleni Kastriti; Julian Petersen; Tatiana Chontorotzea; Lukas Englmaier; Natalia Akkuratova; Yunshi Yang; Martin Häring; Viacheslav Dyachuk; et al.Christoph BockMatthias FarlikMichael L. PiacentinoFranck BoismoreauMarkus M. HilscherChika YokotaXiaoyan QianMats NilssonMarianne E. BronnerLaura CrociWen-Yu HsiaoDavid A. GuertinJean-Francois BrunetGian Giacomo ConsalezPatrik ErnforsKaj FriedPeter V. KharchenkoIgor Adameyko Spatiotemporal structure of cell fate decisions in murine neural crest. Science 2019, 364, eaas9536, 10.1126/science.aas9536.
- Jennifer Soto; Xili Ding; Aijun Wang; Song Li; Neural crest-like stem cells for tissue regeneration. Stem Cells Translational Medicine 2021, 10, 681-693, 10.1002/sctm.20-0361.
- Jon Golding; James Cohen; Border Controls at the Mammalian Spinal Cord: Late-Surviving Neural Crest Boundary Cap Cells at Dorsal Root Entry Sites May Regulate Sensory Afferent Ingrowth and Entry Zone Morphogenesis. Molecular and Cellular Neuroscience 1997, 9, 381-396, 10.1006/mcne.1997.0647.
- C. Niederlander; A. Lumsden; Late emigrating neural crest cells migrate specifically to the exit points of cranial branchiomotor nerves. Development 1996, 122, 2367-2374, 10.1242/dev.122.8.2367.
- Katarzyna J .Radomska; Piotr Topilko; Boundary cap cells in development and disease. Current Opinion in Neurobiology 2017, 47, 209-215, 10.1016/j.conb.2017.11.003.
- Piotr Topilko; Sylvie Schneider-Maunoury; Giovanni Levi; Anne Baron-Van Evercooren; Amina Ben Younes Chennoufi; Tania Seitanidou; Charles Babinet; Patrick Charnay; Krox-20 controls myelination in the peripheral nervous system. Nature Cell Biology 1994, 371, 796-799, 10.1038/371796a0.
- Violetta Zujovic; Julie Thibaud; Corinne Bachelin; Marie Vidal; Cyrille Deboux; Fanny Coulpier; Nicolas Stadler; Patrick Charnay; Piotr Topilko; Anne Baron-Van Evercooren; et al. Boundary cap cells are peripheral nervous system stem cells that can be redirected into central nervous system lineages. Proceedings of the National Academy of Sciences 2011, 108, 10714-10719, 10.1073/pnas.1018687108.
- Aurélie Gresset; Fanny Coulpier; Gaspard Gerschenfeld; Alexandre Jourdon; Graziella Matesic; Laurence Richard; Jean-Michel Vallat; Patrick Charnay; Piotr Topilko; Boundary Caps Give Rise to Neurogenic Stem Cells and Terminal Glia in the Skin. Stem Cell Reports 2015, 5, 278-290, 10.1016/j.stemcr.2015.06.005.
- Fanny Coulpier; Stéphane Le Crom; Géraldine S. Maro; Jan Manent; Marco Giovannini; Zofia Maciorowski; Andreas Fischer; Manfred Gessler; Patrick Charnay; Piotr Topilko; et al. Novel features of boundary cap cells revealed by the analysis of newly identified molecular markers. Glia 2009, 57, 1450-1457, 10.1002/glia.20862.
- Ziping Dong; Andrea Sinanan; David Parkinson; Eric Parmantier; Rhona Mirsky; Kristján R. Jessen; Schwann cell development in embryonic mouse nerves. Journal of Neuroscience Research 1999, 56, 334-348, 10.1002/(sici)1097-4547(19990515)56:4<334::aid-jnr2>3.3.co;2-r.
- Kristjan R. Jessen; Rhona Mirsky; The origin and development of glial cells in peripheral nerves. Nature Reviews Neuroscience 2005, 6, 671-682, 10.1038/nrn1746.
- Géraldine S Maro; Matthieu Vermeren; Octavian Voiculescu; Lisa Melton; James Cohen; Patrick Charnay; Piotr Topilko; Neural crest boundary cap cells constitute a source of neuronal and glial cells of the PNS. Nature Neuroscience 2004, 7, 930-938, 10.1038/nn1299.
- Kristjan R Jessen; Rhona Mirsky; Schwann cells and their precursors emerge as major regulators of nerve development. Trends in Neurosciences 1999, 22, 402-410, 10.1016/s0166-2236(98)01391-5.
- Kristjan R. Jessen; Rhona Mirsky; Schwann Cell Precursors; Multipotent Glial Cells in Embryonic Nerves. Frontiers in Molecular Neuroscience 2019, 12, 69, 10.3389/fnmol.2019.00069.
- Jennifer Soto; Paula Monje; Axon contact-driven Schwann cell dedifferentiation.. Glia 2017, 65, 864-882, 10.1002/glia.23131.
- M. Laura Feltri; Yannick Poitelon; Stefano Carlo Previtali; How Schwann Cells Sort Axons. The Neuroscientist 2015, 22, 252-265, 10.1177/1073858415572361.
- Yuan Zhu; Luis F. Parada; The Molecular and Genetic Basis of Neurological Tumours. Nature Cancer 2002, 2, 616-626, 10.1038/nrc866.
- Yuan Zhu; Pritam Ghosh; Patrick Charnay; Dennis K. Burns; Luis F. Parada; Neurofibromas in NF1: Schwann Cell Origin and Role of Tumor Environment. Science 2002, 296, 920-922, 10.1126/science.1068452.
- Nancy M. Joseph; Jack T. Mosher; Johanna Buchstaller; Paige Snider; Paul E. McKeever; Megan Lim; Simon J. Conway; Luis F. Parada; Yuan Zhu; Sean J. Morrison; et al. The Loss of Nf1 Transiently Promotes Self-Renewal but Not Tumorigenesis by Neural Crest Stem Cells. Cancer Cell 2008, 13, 129-140, 10.1016/j.ccr.2008.01.003.
- Huarui Zheng; Lou Chang; Neha Patel; Jiong Yang; Lori Lowe; Dennis K. Burns; Yuan Zhu; Induction of Abnormal Proliferation by Nonmyelinating Schwann Cells Triggers Neurofibroma Formation. Cancer Cell 2008, 13, 117-128, 10.1016/j.ccr.2008.01.002.
- H Saito; T Yoshida; H Yamazaki; N Suzuki; Conditional N-rasG12V expression promotes manifestations of neurofibromatosis in a mouse model. Oncogene 2007, 26, 4714-4719, 10.1038/sj.onc.1210250.
- Jianqiang Wu; Jon P. Williams; Tilat A. Rizvi; Jennifer J. Kordich; David Witte; Dies Meijer; Anat O. Stemmer-Rachamimov; Jose A. Cancelas; Nancy Ratner; Plexiform and Dermal Neurofibromas and Pigmentation Are Caused by Nf1 Loss in Desert Hedgehog-Expressing Cells. Cancer Cell 2008, 13, 105-116, 10.1016/j.ccr.2007.12.027.
- Lu Q. Le; Tracey Shipman; Dennis K. Burns; Luis F. Parada; Cell of Origin and Microenvironment Contribution for NF1-Associated Dermal Neurofibromas. Cell Stem Cell 2009, 4, 453-463, 10.1016/j.stem.2009.03.017.
- Zhiguo Chen; Juan Mo; Jean-Philippe Brosseau; Tracey Shipman; Yong Wang; Chung-Ping Liao; Jonathan M. Cooper; Robert J. Allaway; Sara J.C. Gosline; Justin Guinney; et al.Thomas J. CarrollLu Q. Le Spatiotemporal Loss of NF1 in Schwann Cell Lineage Leads to Different Types of Cutaneous Neurofibroma Susceptible to Modification by the Hippo Pathway. Cancer Discovery 2019, 9, 114-129, 10.1158/2159-8290.cd-18-0151.
- Katarzyna J. Radomska; Fanny Coulpier; Aurelie Gresset; Alain Schmitt; Amal Debbiche; Sophie Lemoine; Pierre Wolkenstein; Jean-Michel Vallat; Patrick Charnay; Piotr Topilko; et al. Cellular Origin, Tumor Progression, and Pathogenic Mechanisms of Cutaneous Neurofibromas Revealed by Mice with Nf1 Knockout in Boundary Cap Cells. Cancer Discovery 2019, 9, 130-147, 10.1158/2159-8290.cd-18-0156.
- Juan Mo; Corina Anastasaki; Zhiguo Chen; Tracey Shipman; Jason B. Papke; Kevin Y. Yin; David H. Gutmann; Lu Q. Le; Humanized neurofibroma model from induced pluripotent stem cells delineates tumor pathogenesis and developmental origins. Journal of Clinical Investigation 2021, 131, e139807, 10.1172/jci139807.
- Gabriel Corfas; Miguel Omar Velardez; Chien-Ping Ko; Nancy Ratner; Elior Peles; Mechanisms and Roles of Axon-Schwann Cell Interactions. The Journal of Neuroscience 2004, 24, 9250-9260, 10.1523/jneurosci.3649-04.2004.
- Simona Parrinello; Luke A. Noon; Marie C. Harrisingh; Patrick Wingfield Digby; Laura H. Rosenberg; Catherine A. Cremona; Pedro Echave; Adrienne M. Flanagan; Luis F. Parada; Alison C. Lloyd; et al. NF1 loss disrupts Schwann cell–axonal interactions: a novel role for semaphorin 4F. Genes & Development 2008, 22, 3335-3348, 10.1101/gad.490608.
- Mark S Stonecypher; Stephanie J Byer; William E Grizzle; Steven L Carroll; Activation of the neuregulin-1/ErbB signaling pathway promotes the proliferation of neoplastic Schwann cells in human malignant peripheral nerve sheath tumors. Oncogene 2005, 24, 5589-5605, 10.1038/sj.onc.1208730.
- Simona Parrinello; Alison C. Lloyd; Neurofibroma development in NF1 – insights into tumour initiation. Trends in Cell Biology 2009, 19, 395-403, 10.1016/j.tcb.2009.05.003.
- Simona Parrinello; Alison C. Lloyd; Neurofibroma development in NF1 – insights into tumour initiation. Trends in Cell Biology 2009, 19, 395-403, 10.1016/j.tcb.2009.05.003.
- K. R. Jessen; R. Mirsky; The repair Schwann cell and its function in regenerating nerves. The Journal of Physiology 2016, 594, 3521-3531, 10.1113/jp270874.
- Shlomit Shamash; Fanny Reichert; Shlomo Rotshenker; The Cytokine Network of Wallerian Degeneration: Tumor Necrosis Factor-α, Interleukin-1α, and Interleukin-1β. The Journal of Neuroscience 2002, 22, 3052-3060, 10.1523/jneurosci.22-08-03052.2002.
- Tilat A. Rizvi; Yuan Huang; Amer Sidani; Radhika Atit; David A. Largaespada; Raymond E. Boissy; Nancy Ratner; A novel cytokine pathway suppresses glial cell melanogenesis after injury to adult nerve.. The Journal of Neuroscience 2002, 22, 9831-40, 10.1523/jneurosci.22-22-09831.2002.
- Sara Ribeiro; Ilaria Napoli; Ian J. White; Simona Parrinello; Adrienne M. Flanagan; Ueli Suter; Luis F. Parada; Alison C. Lloyd; Injury Signals Cooperate with Nf1 Loss to Relieve the Tumor-Suppressive Environment of Adult Peripheral Nerve. Cell Reports 2013, 5, 126-136, 10.1016/j.celrep.2013.08.033.
- Adam J. Singer; Richard A.F. Clark; Cutaneous Wound Healing. New England Journal of Medicine 1999, 341, 738-746, 10.1056/nejm199909023411006.
- Simona Parrinello; Alison C. Lloyd; Neurofibroma development in NF1 – insights into tumour initiation. Trends in Cell Biology 2009, 19, 395-403, 10.1016/j.tcb.2009.05.003.
- Simona Parrinello; Alison C. Lloyd; Neurofibroma development in NF1 – insights into tumour initiation. Trends in Cell Biology 2009, 19, 395-403, 10.1016/j.tcb.2009.05.003.
- Chung-Ping Liao; Reid C. Booker; Jean-Philippe Brosseau; Zhiguo Chen; Juan Mo; Edem Tchegnon; Yong Wang; D. Wade Clapp; Lu Q. Le; Contributions of inflammation and tumor microenvironment to neurofibroma tumorigenesis. Journal of Clinical Investigation 2018, 128, 2848-2861, 10.1172/jci99424.
- Feng-Chun Yang; David A. Ingram; Shi Chen; Yuan Zhu; Jin Yuan; Xiaohong Li; Xianlin Yang; Scott Knowles; Whitney Horn; Yan Li; et al.Shaobo ZhangYanzhu YangSaeed T. VakiliMenggang YuDennis BurnsKent RobertsonGary HutchinsLuis F. ParadaD. Wade Clapp Nf1-Dependent Tumors Require a Microenvironment Containing Nf1+/−- and c-kit-Dependent Bone Marrow. Cell 2008, 135, 437-448, 10.1016/j.cell.2008.08.041.
- Fang Li; Amy M. Munchhof; Hilary A. White; Laura E. Mead; Theresa R. Krier; Amy Fenoglio; Shi Chen; Xiaohua Wu; Shanbao Cai; Feng-Chun Yang; et al.David A. Ingram Neurofibromin is a novel regulator of RAS-induced signals in primary vascular smooth muscle cells. Human Molecular Genetics 2006, 15, 1921-1930, 10.1093/hmg/ddl114.
- Karl Staser; Feng-Chun Yang; D. Wade Clapp; Pathogenesis of Plexiform Neurofibroma: Tumor-Stromal/Hematopoietic Interactions in Tumor Progression. Annual Review of Pathology: Mechanisms of Disease 2012, 7, 469-495, 10.1146/annurev-pathol-011811-132441.
- Rafaela E. Rozza-De-Menezes; Nicolle Cavalcante Gaglionone; Raquel M. Andrade-Losso; Orlando H. K. Siqueira; Lilian M. Almeida; Kamila Da S. Peruzini; Marco A. C. Guimarães-Filho; Carolina I. Brum; Mauro Geller; Karin S. Cunha; et al. Receptor of ghrelin is expressed in cutaneous neurofibromas of individuals with neurofibromatosis 1. Orphanet Journal of Rare Diseases 2017, 12, 186, 10.1186/s13023-017-0734-x.
More
Information
Subjects:
Health Care Sciences & Services
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
890
Revisions:
4 times
(View History)
Update Date:
26 Sep 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No