Neurofibromatosis type 1 (NF1), a genetic tumor predisposition syndrome that affects about 1 in 3000 newborns, is caused by mutations in the NF1 gene and subsequent inactivation of its encoded neurofibromin. Neurofibromin is a tumor suppressor protein involved in the downregulation of Ras signaling. Despite a diverse clinical spectrum, one of several hallmarks of NF1 is a peripheral nerve sheath tumor (PNST), which comprises mixed nervous and fibrous components. The distinct spatiotemporal characteristics of plexiform and cutaneous neurofibromas have prompted hypotheses about the origin and developmental features of these tumors, involving various cellular transition processes.
- NF1
- neurofibroma formation
- SCPs
1. Neurofibroma Formation
1.1. The Developmental Origin of Schwann Cell Lineages
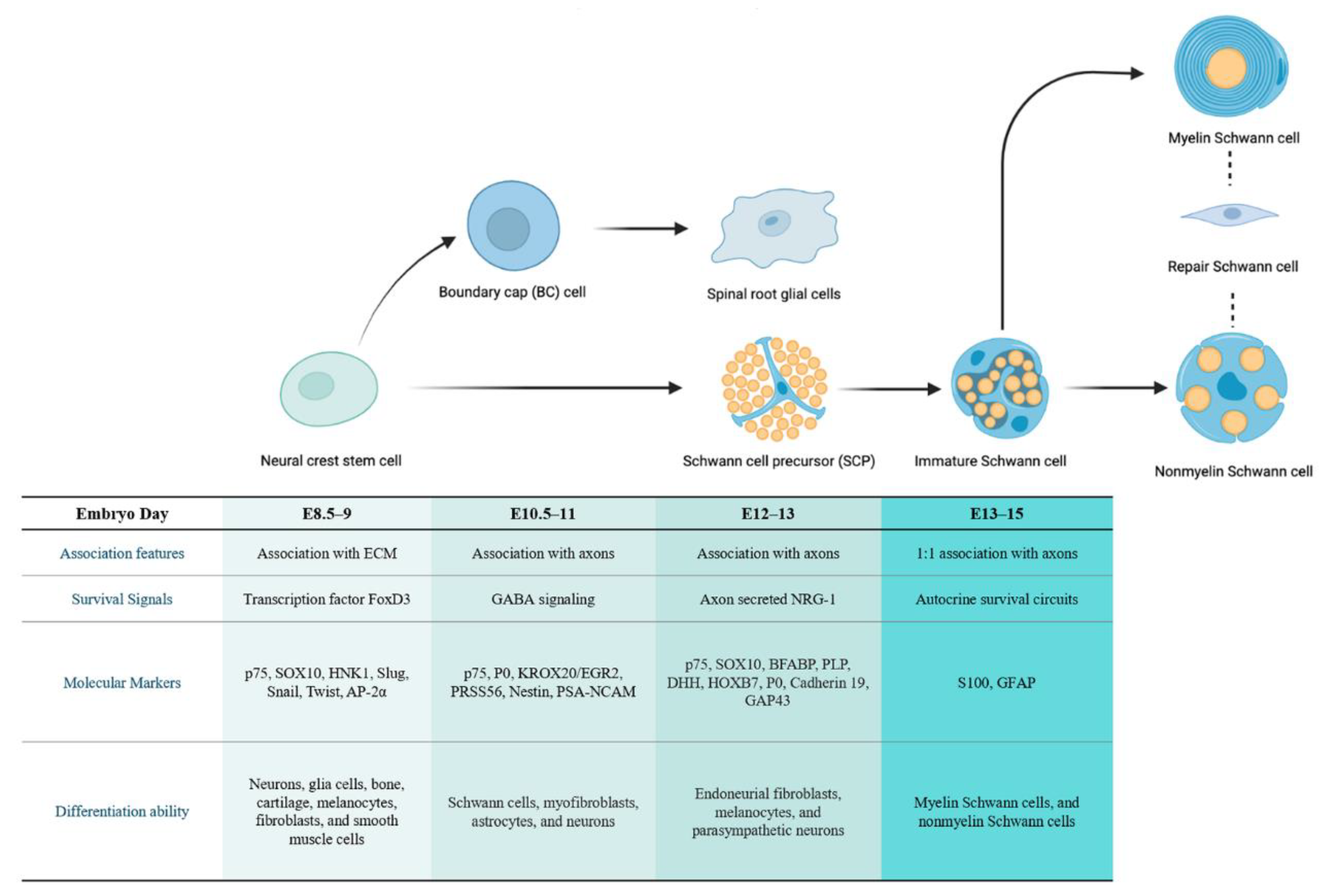
1.1.1. The Cellular Origin of Neurofibroma
1.1.2. The Cellular Origin of pNF
1.1.3. The Cellular Origin of cNF
1.1.4. Associate pNF and cNF with a Common Stage of Origin
1.2. Alterations in Schwann Cells in the Early Stage of Tumorigenesis
2. Neurofibroma Progression
2.1. Schwann Cells Contribution and Lineage Shift
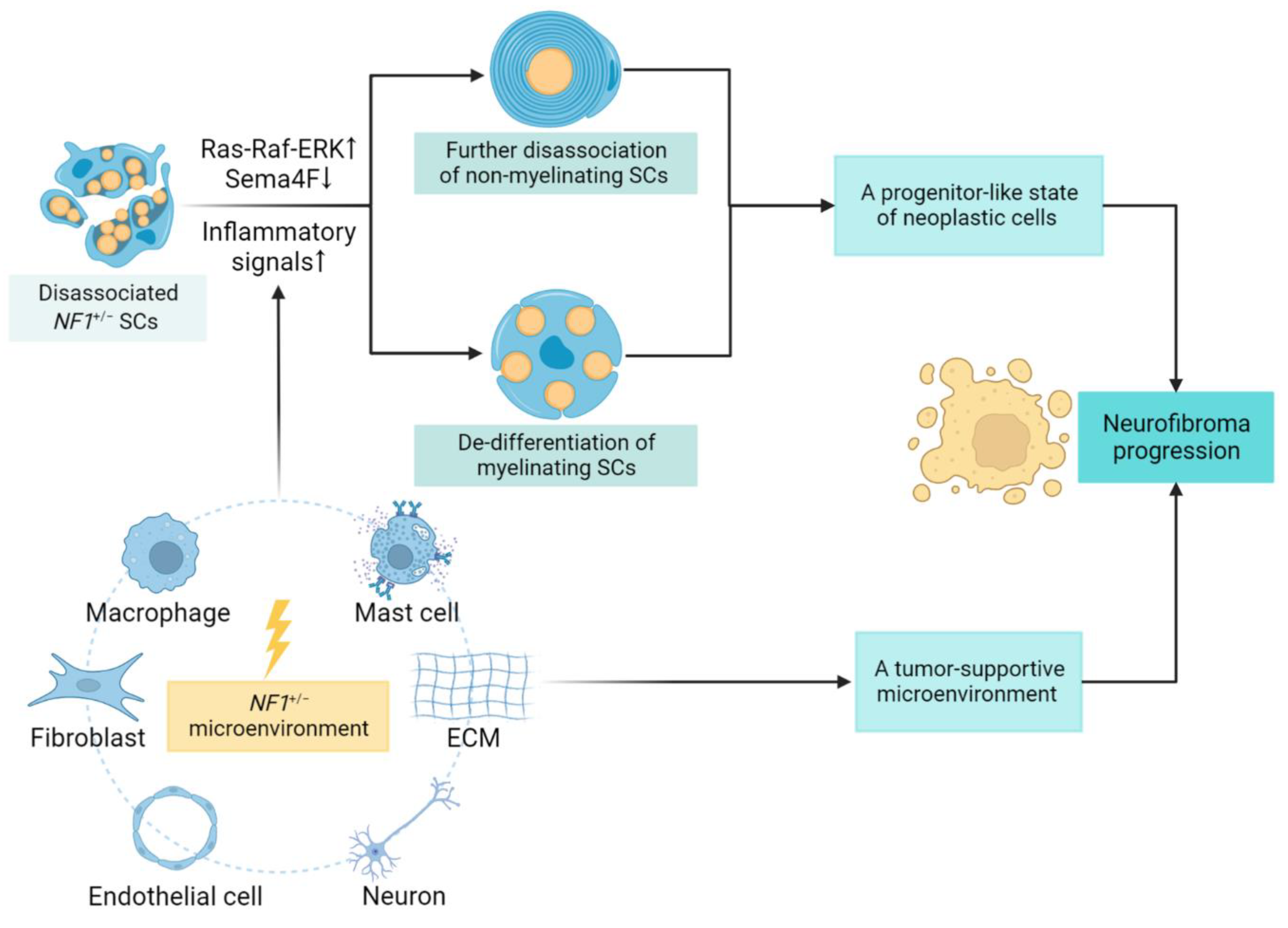
2.2. Role of the Tumor Microenvironment
This entry is adapted from the peer-reviewed paper 10.3390/cancers14184513
References
- Crump, T.; Translation of case reports in Ueber die multiplen Fibrome der Haut und ihre Beziehung zu den multiplen Neuromen by F. v. Recklinghausen.. Advances in Neurology 1981, 29, 259-275, .
- Yoshimasa Kamata; Study on the ultrastructure and acetylcholinesterase activity in von Recklinghausen's neurofibromatosis. Pathology International 1978, 28, 393-410, 10.1111/j.1440-1827.1978.tb01264.x.
- Lu Q. Le; Chiachi Liu; Tracey Shipman; Zhiguo Chen; Ueli Suter; Luis F. Parada; Susceptible Stages in Schwann Cells for NF1-Associated Plexiform Neurofibroma Development. Cancer Research 2011, 71, 4686-4695, 10.1158/0008-5472.can-10-4577.
- Debra A. Mayes; Tilat A. Rizvi; Jose A. Cancelas; Nathan T. Kolasinski; Georgianne M. Ciraolo; Anat O. Stemmer-Rachamimov; Nancy Ratner; Perinatal or adult Nf1 inactivation using tamoxifen-inducible PlpCre each cause neurofibroma formation. Cancer Research 2011, 71, 4675-4685, 10.1158/0008-5472.can-10-4558.
- Vincent W. Keng; Eric P. Rahrmann; Adrienne L. Watson; Barbara R. Tschida; Christopher L. Moertel; Walter J. Jessen; Tilat A. Rizvi; Margaret H. Collins; Nancy Ratner; David A. Largaespada; et al. PTEN and NF1 inactivation in Schwann cells produces a severe phenotype in the peripheral nervous system that promotes the development and malignant progression of peripheral nerve sheath tumors. Cancer Research 2012, 72, 3405-3413, 10.1158/0008-5472.can-11-4092.
- Zhiguo Chen; Chiachi Liu; Amish Patel; Chung-Ping Liao; Yong Wang; Lu Q. Le; Cells of Origin in the Embryonic Nerve Roots for NF1-Associated Plexiform Neurofibroma. Cancer Cell 2014, 26, 695-706, 10.1016/j.ccell.2014.09.009.
- Katherine E. Chaney; Melissa R. Perrino; Leah J. Kershner; Ami V. Patel; Jianqiang Wu; Kwangmin Choi; Tilat A. Rizvi; Eva Dombi; Sara Szabo; David A. Largaespada; et al. Cdkn2a Loss in a Model of Neurofibroma Demonstrates Stepwise Tumor Progression to Atypical Neurofibroma and MPNST. Cancer Research 2020, 80, 4720-4730, 10.1158/0008-5472.can-19-1429.
- Derek L. Stemple; David J. Anderson; Isolation of a stem cell for neurons and glia from the mammalian neural crest. Cell 1992, 71, 973-985, 10.1016/0092-8674(92)90393-q.
- Annita Achilleos; Paul A Trainor; Neural crest stem cells: discovery, properties and potential for therapy. Cell Research 2012, 22, 288-304, 10.1038/cr.2012.11.
- Ruslan Soldatov; Marketa Kaucka; Maria Eleni Kastriti; Julian Petersen; Tatiana Chontorotzea; Lukas Englmaier; Natalia Akkuratova; Yunshi Yang; Martin Häring; Viacheslav Dyachuk; et al. Spatiotemporal structure of cell fate decisions in murine neural crest. Science 2019, 364, eaas9536, 10.1126/science.aas9536.
- Jennifer Soto; Xili Ding; Aijun Wang; Song Li; Neural crest-like stem cells for tissue regeneration. Stem Cells Translational Medicine 2021, 10, 681-693, 10.1002/sctm.20-0361.
- Jon Golding; James Cohen; Border Controls at the Mammalian Spinal Cord: Late-Surviving Neural Crest Boundary Cap Cells at Dorsal Root Entry Sites May Regulate Sensory Afferent Ingrowth and Entry Zone Morphogenesis. Molecular and Cellular Neuroscience 1997, 9, 381-396, 10.1006/mcne.1997.0647.
- C. Niederlander; A. Lumsden; Late emigrating neural crest cells migrate specifically to the exit points of cranial branchiomotor nerves. Development 1996, 122, 2367-2374, 10.1242/dev.122.8.2367.
- Katarzyna J .Radomska; Piotr Topilko; Boundary cap cells in development and disease. Current Opinion in Neurobiology 2017, 47, 209-215, 10.1016/j.conb.2017.11.003.
- Piotr Topilko; Sylvie Schneider-Maunoury; Giovanni Levi; Anne Baron-Van Evercooren; Amina Ben Younes Chennoufi; Tania Seitanidou; Charles Babinet; Patrick Charnay; Krox-20 controls myelination in the peripheral nervous system. Nature Cell Biology 1994, 371, 796-799, 10.1038/371796a0.
- Violetta Zujovic; Julie Thibaud; Corinne Bachelin; Marie Vidal; Cyrille Deboux; Fanny Coulpier; Nicolas Stadler; Patrick Charnay; Piotr Topilko; Anne Baron-Van Evercooren; et al. Boundary cap cells are peripheral nervous system stem cells that can be redirected into central nervous system lineages. Proceedings of the National Academy of Sciences 2011, 108, 10714-10719, 10.1073/pnas.1018687108.
- Aurélie Gresset; Fanny Coulpier; Gaspard Gerschenfeld; Alexandre Jourdon; Graziella Matesic; Laurence Richard; Jean-Michel Vallat; Patrick Charnay; Piotr Topilko; Boundary Caps Give Rise to Neurogenic Stem Cells and Terminal Glia in the Skin. Stem Cell Reports 2015, 5, 278-290, 10.1016/j.stemcr.2015.06.005.
- Fanny Coulpier; Stéphane Le Crom; Géraldine S. Maro; Jan Manent; Marco Giovannini; Zofia Maciorowski; Andreas Fischer; Manfred Gessler; Patrick Charnay; Piotr Topilko; et al. Novel features of boundary cap cells revealed by the analysis of newly identified molecular markers. Glia 2009, 57, 1450-1457, 10.1002/glia.20862.
- Ziping Dong; Andrea Sinanan; David Parkinson; Eric Parmantier; Rhona Mirsky; Kristján R. Jessen; Schwann cell development in embryonic mouse nerves. Journal of Neuroscience Research 1999, 56, 334-348, 10.1002/(sici)1097-4547(19990515)56:4<334::aid-jnr2>3.3.co;2-r.
- Kristjan R. Jessen; Rhona Mirsky; The origin and development of glial cells in peripheral nerves. Nature Reviews Neuroscience 2005, 6, 671-682, 10.1038/nrn1746.
- Géraldine S Maro; Matthieu Vermeren; Octavian Voiculescu; Lisa Melton; James Cohen; Patrick Charnay; Piotr Topilko; Neural crest boundary cap cells constitute a source of neuronal and glial cells of the PNS. Nature Neuroscience 2004, 7, 930-938, 10.1038/nn1299.
- Kristjan R Jessen; Rhona Mirsky; Schwann cells and their precursors emerge as major regulators of nerve development. Trends in Neurosciences 1999, 22, 402-410, 10.1016/s0166-2236(98)01391-5.
- Kristjan R. Jessen; Rhona Mirsky; Schwann Cell Precursors; Multipotent Glial Cells in Embryonic Nerves. Frontiers in Molecular Neuroscience 2019, 12, 69, 10.3389/fnmol.2019.00069.
- Jennifer Soto; Paula Monje; Axon contact-driven Schwann cell dedifferentiation.. Glia 2017, 65, 864-882, 10.1002/glia.23131.
- M. Laura Feltri; Yannick Poitelon; Stefano Carlo Previtali; How Schwann Cells Sort Axons. The Neuroscientist 2015, 22, 252-265, 10.1177/1073858415572361.
- Yuan Zhu; Luis F. Parada; The Molecular and Genetic Basis of Neurological Tumours. Nature Cancer 2002, 2, 616-626, 10.1038/nrc866.
- Yuan Zhu; Pritam Ghosh; Patrick Charnay; Dennis K. Burns; Luis F. Parada; Neurofibromas in NF1: Schwann Cell Origin and Role of Tumor Environment. Science 2002, 296, 920-922, 10.1126/science.1068452.
- Nancy M. Joseph; Jack T. Mosher; Johanna Buchstaller; Paige Snider; Paul E. McKeever; Megan Lim; Simon J. Conway; Luis F. Parada; Yuan Zhu; Sean J. Morrison; et al. The Loss of Nf1 Transiently Promotes Self-Renewal but Not Tumorigenesis by Neural Crest Stem Cells. Cancer Cell 2008, 13, 129-140, 10.1016/j.ccr.2008.01.003.
- Huarui Zheng; Lou Chang; Neha Patel; Jiong Yang; Lori Lowe; Dennis K. Burns; Yuan Zhu; Induction of Abnormal Proliferation by Nonmyelinating Schwann Cells Triggers Neurofibroma Formation. Cancer Cell 2008, 13, 117-128, 10.1016/j.ccr.2008.01.002.
- H Saito; T Yoshida; H Yamazaki; N Suzuki; Conditional N-rasG12V expression promotes manifestations of neurofibromatosis in a mouse model. Oncogene 2007, 26, 4714-4719, 10.1038/sj.onc.1210250.
- Jianqiang Wu; Jon P. Williams; Tilat A. Rizvi; Jennifer J. Kordich; David Witte; Dies Meijer; Anat O. Stemmer-Rachamimov; Jose A. Cancelas; Nancy Ratner; Plexiform and Dermal Neurofibromas and Pigmentation Are Caused by Nf1 Loss in Desert Hedgehog-Expressing Cells. Cancer Cell 2008, 13, 105-116, 10.1016/j.ccr.2007.12.027.
- Lu Q. Le; Tracey Shipman; Dennis K. Burns; Luis F. Parada; Cell of Origin and Microenvironment Contribution for NF1-Associated Dermal Neurofibromas. Cell Stem Cell 2009, 4, 453-463, 10.1016/j.stem.2009.03.017.
- Zhiguo Chen; Juan Mo; Jean-Philippe Brosseau; Tracey Shipman; Yong Wang; Chung-Ping Liao; Jonathan M. Cooper; Robert J. Allaway; Sara J.C. Gosline; Justin Guinney; et al. Spatiotemporal Loss of NF1 in Schwann Cell Lineage Leads to Different Types of Cutaneous Neurofibroma Susceptible to Modification by the Hippo Pathway. Cancer Discovery 2019, 9, 114-129, 10.1158/2159-8290.cd-18-0151.
- Katarzyna J. Radomska; Fanny Coulpier; Aurelie Gresset; Alain Schmitt; Amal Debbiche; Sophie Lemoine; Pierre Wolkenstein; Jean-Michel Vallat; Patrick Charnay; Piotr Topilko; et al. Cellular Origin, Tumor Progression, and Pathogenic Mechanisms of Cutaneous Neurofibromas Revealed by Mice with Nf1 Knockout in Boundary Cap Cells. Cancer Discovery 2019, 9, 130-147, 10.1158/2159-8290.cd-18-0156.
- Juan Mo; Corina Anastasaki; Zhiguo Chen; Tracey Shipman; Jason B. Papke; Kevin Y. Yin; David H. Gutmann; Lu Q. Le; Humanized neurofibroma model from induced pluripotent stem cells delineates tumor pathogenesis and developmental origins. Journal of Clinical Investigation 2021, 131, e139807, 10.1172/jci139807.
- Gabriel Corfas; Miguel Omar Velardez; Chien-Ping Ko; Nancy Ratner; Elior Peles; Mechanisms and Roles of Axon-Schwann Cell Interactions. The Journal of Neuroscience 2004, 24, 9250-9260, 10.1523/jneurosci.3649-04.2004.
- Simona Parrinello; Luke A. Noon; Marie C. Harrisingh; Patrick Wingfield Digby; Laura H. Rosenberg; Catherine A. Cremona; Pedro Echave; Adrienne M. Flanagan; Luis F. Parada; Alison C. Lloyd; et al. NF1 loss disrupts Schwann cell–axonal interactions: a novel role for semaphorin 4F. Genes & Development 2008, 22, 3335-3348, 10.1101/gad.490608.
- Mark S Stonecypher; Stephanie J Byer; William E Grizzle; Steven L Carroll; Activation of the neuregulin-1/ErbB signaling pathway promotes the proliferation of neoplastic Schwann cells in human malignant peripheral nerve sheath tumors. Oncogene 2005, 24, 5589-5605, 10.1038/sj.onc.1208730.
- Simona Parrinello; Alison C. Lloyd; Neurofibroma development in NF1 – insights into tumour initiation. Trends in Cell Biology 2009, 19, 395-403, 10.1016/j.tcb.2009.05.003.
- Simona Parrinello; Alison C. Lloyd; Neurofibroma development in NF1 – insights into tumour initiation. Trends in Cell Biology 2009, 19, 395-403, 10.1016/j.tcb.2009.05.003.
- K. R. Jessen; R. Mirsky; The repair Schwann cell and its function in regenerating nerves. The Journal of Physiology 2016, 594, 3521-3531, 10.1113/jp270874.
- Shlomit Shamash; Fanny Reichert; Shlomo Rotshenker; The Cytokine Network of Wallerian Degeneration: Tumor Necrosis Factor-α, Interleukin-1α, and Interleukin-1β. The Journal of Neuroscience 2002, 22, 3052-3060, 10.1523/jneurosci.22-08-03052.2002.
- Tilat A. Rizvi; Yuan Huang; Amer Sidani; Radhika Atit; David A. Largaespada; Raymond E. Boissy; Nancy Ratner; A novel cytokine pathway suppresses glial cell melanogenesis after injury to adult nerve.. The Journal of Neuroscience 2002, 22, 9831-40, 10.1523/jneurosci.22-22-09831.2002.
- Sara Ribeiro; Ilaria Napoli; Ian J. White; Simona Parrinello; Adrienne M. Flanagan; Ueli Suter; Luis F. Parada; Alison C. Lloyd; Injury Signals Cooperate with Nf1 Loss to Relieve the Tumor-Suppressive Environment of Adult Peripheral Nerve. Cell Reports 2013, 5, 126-136, 10.1016/j.celrep.2013.08.033.
- Adam J. Singer; Richard A.F. Clark; Cutaneous Wound Healing. New England Journal of Medicine 1999, 341, 738-746, 10.1056/nejm199909023411006.
- Simona Parrinello; Alison C. Lloyd; Neurofibroma development in NF1 – insights into tumour initiation. Trends in Cell Biology 2009, 19, 395-403, 10.1016/j.tcb.2009.05.003.
- Simona Parrinello; Alison C. Lloyd; Neurofibroma development in NF1 – insights into tumour initiation. Trends in Cell Biology 2009, 19, 395-403, 10.1016/j.tcb.2009.05.003.
- Chung-Ping Liao; Reid C. Booker; Jean-Philippe Brosseau; Zhiguo Chen; Juan Mo; Edem Tchegnon; Yong Wang; D. Wade Clapp; Lu Q. Le; Contributions of inflammation and tumor microenvironment to neurofibroma tumorigenesis. Journal of Clinical Investigation 2018, 128, 2848-2861, 10.1172/jci99424.
- Feng-Chun Yang; David A. Ingram; Shi Chen; Yuan Zhu; Jin Yuan; Xiaohong Li; Xianlin Yang; Scott Knowles; Whitney Horn; Yan Li; et al. Nf1-Dependent Tumors Require a Microenvironment Containing Nf1+/−- and c-kit-Dependent Bone Marrow. Cell 2008, 135, 437-448, 10.1016/j.cell.2008.08.041.
- Fang Li; Amy M. Munchhof; Hilary A. White; Laura E. Mead; Theresa R. Krier; Amy Fenoglio; Shi Chen; Xiaohua Wu; Shanbao Cai; Feng-Chun Yang; et al. Neurofibromin is a novel regulator of RAS-induced signals in primary vascular smooth muscle cells. Human Molecular Genetics 2006, 15, 1921-1930, 10.1093/hmg/ddl114.
- Karl Staser; Feng-Chun Yang; D. Wade Clapp; Pathogenesis of Plexiform Neurofibroma: Tumor-Stromal/Hematopoietic Interactions in Tumor Progression. Annual Review of Pathology: Mechanisms of Disease 2012, 7, 469-495, 10.1146/annurev-pathol-011811-132441.
- Rafaela E. Rozza-De-Menezes; Nicolle Cavalcante Gaglionone; Raquel M. Andrade-Losso; Orlando H. K. Siqueira; Lilian M. Almeida; Kamila Da S. Peruzini; Marco A. C. Guimarães-Filho; Carolina I. Brum; Mauro Geller; Karin S. Cunha; et al. Receptor of ghrelin is expressed in cutaneous neurofibromas of individuals with neurofibromatosis 1. Orphanet Journal of Rare Diseases 2017, 12, 186, 10.1186/s13023-017-0734-x.