 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Brunette Katsandegwaza | -- | 2710 | 2022-09-22 16:29:45 | | | |
| 2 | Lindsay Dong | -51 word(s) | 2659 | 2022-09-26 04:58:17 | | | | |
| 3 | Lindsay Dong | Meta information modification | 2659 | 2022-09-26 08:21:31 | | |
Video Upload Options
Crohn’s disease (CD) and ulcerative colitis (UC) are both highly inflammatory diseases of the gastrointestinal tract, collectively known as inflammatory bowel disease (IBD). Although the cause of IBD is still unclear, several experimental IBD murine models have enabled researchers to make great inroads into understanding human IBD pathology. Murine experimental models of human IBD exhibit immune pathological signatures resembling Crohn’s disease (CD) or ulcerative colitis (UC). These models include the chemical-induced trinitrobenzene sulfonic acid (TNBS) model, oxazolone and dextran sulfate sodium (DSS) models, the gene-deficient I-kappa-B kinase gamma (Iκκ-γ) and interleukin(IL)-10 models, and the CD4+ T-cell transfer model. Although most pre-clinical murine models do not fully recapitulate the complexity of human IBD, these models have added to our knowledge about the causes of disease and have provided targets for developing new treatments.
1. Introduction
2. Chemical Induced Colitis
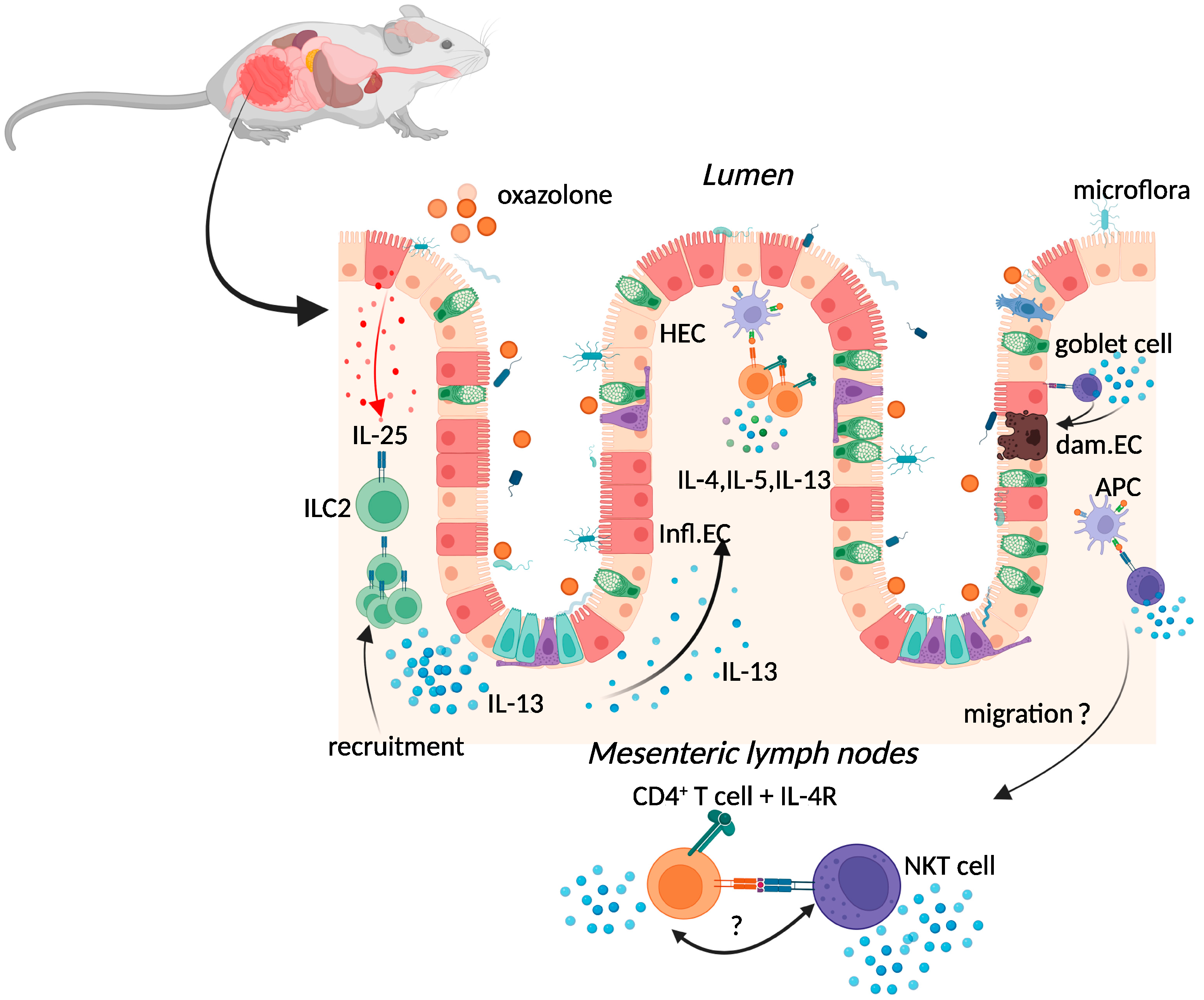
2.1. Oxazolone Colitis
2.2. TNBS-Induced Colitis
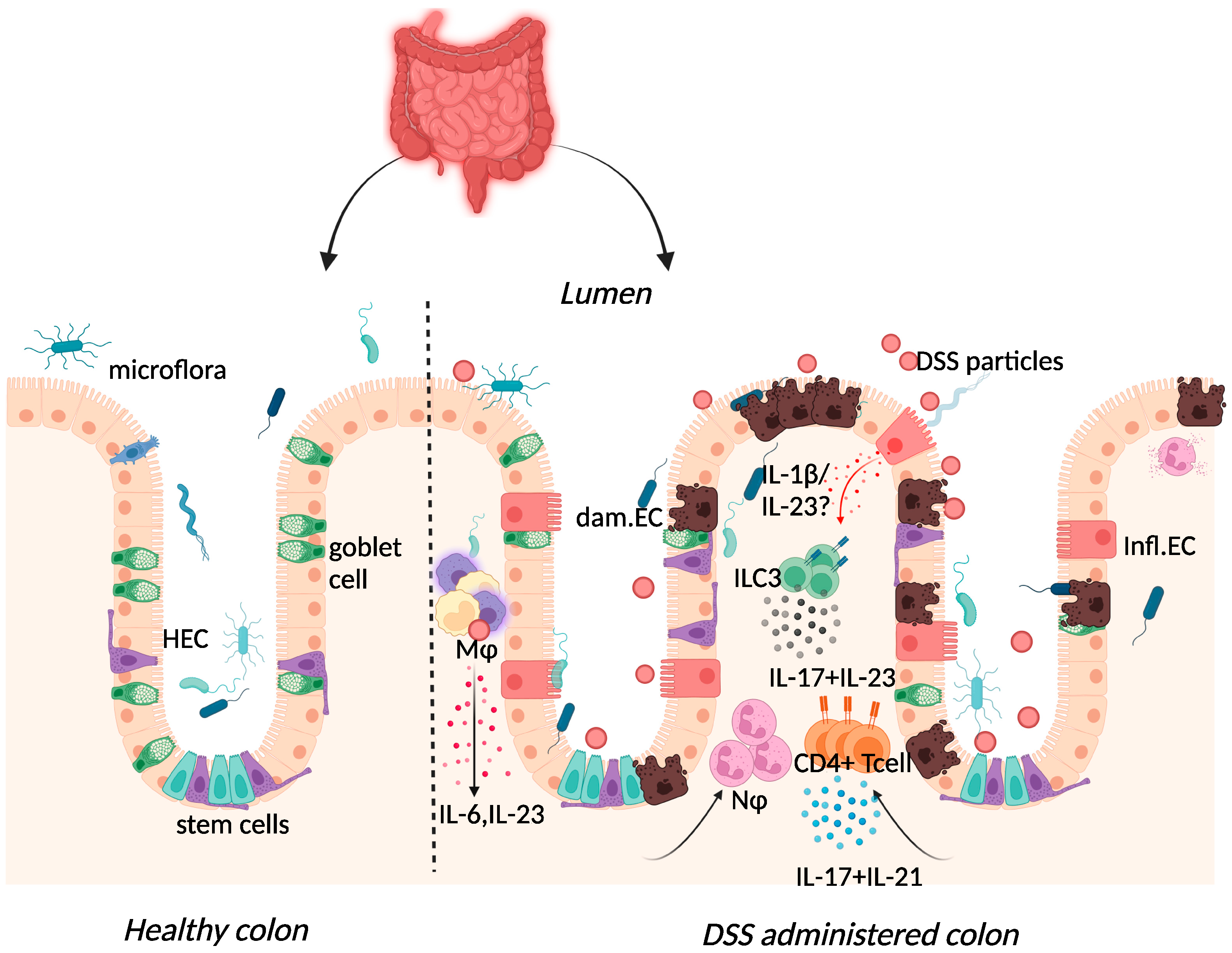
2.3. Dextran-Sulphate-Sodium-Induced Colitis
3. Spontaneous Colitis
3.1. Iκκ-γ (NEMO) Deficiency Colitis
3.2. Interleukin-10 (IL-10) Deficiency Colitis
4. Immune Cell Induced Colitis
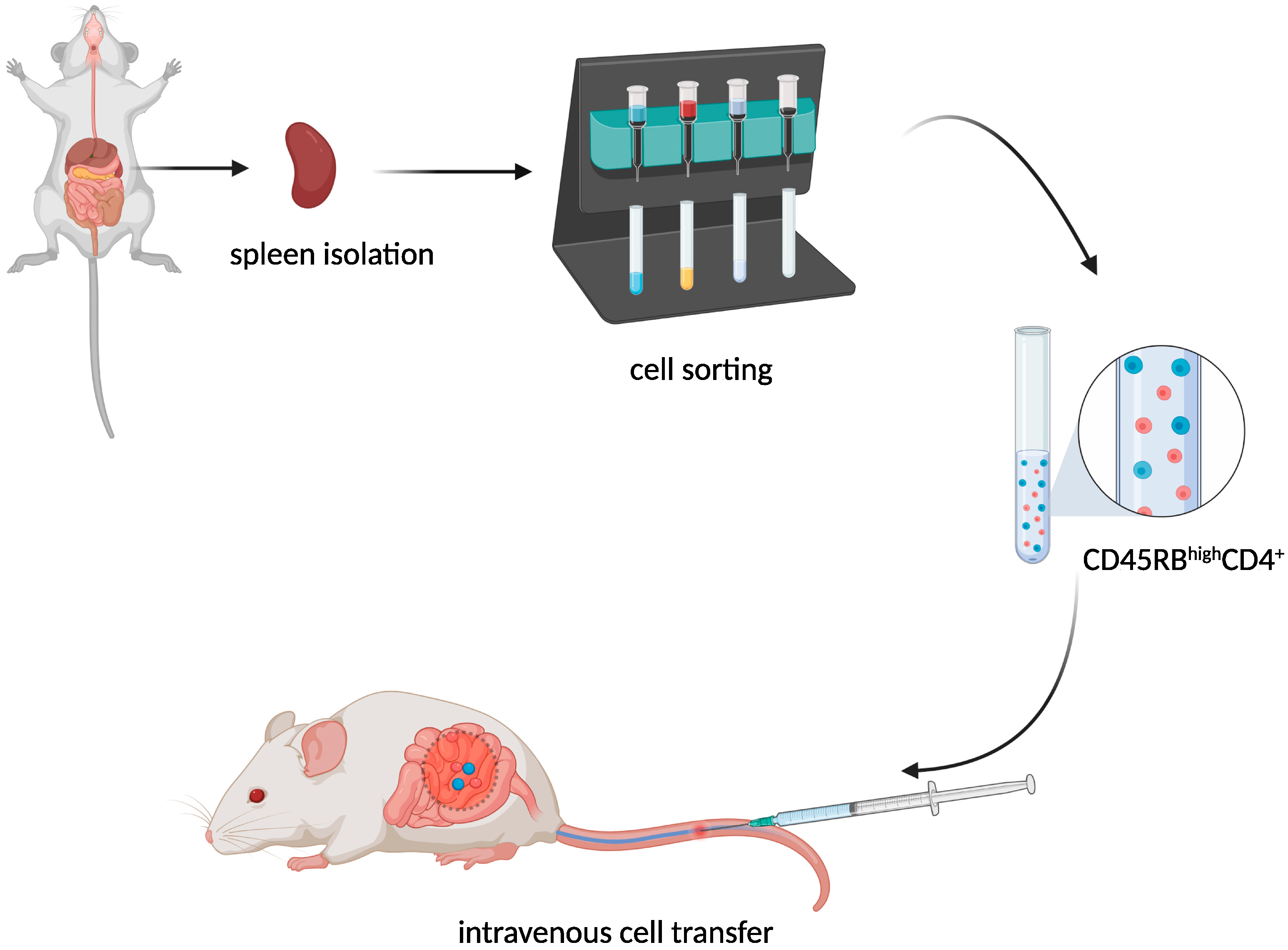
T-Cell Adoptive Transfer Model
References
- Boirivant, M.; Fuss, I.J.; Chu, A.; Strober, W. Oxazolone Colitis: A Murine Model of T Helper Cell Type 2 Colitis Treatable with Antibodies to Interleukin 4. J. Exp. Med. 1998, 188, 1929–1939.
- Kühn, R.; Löhler, J.; Rennick, D.; Rajewsky, K.; Müller, W. Interleukin-10-deficient mice develop chronic enterocolitis. Cell 1993, 75, 263–274.
- Nenci, A.; Becker, C.; Wullaert, A.; Gareus, R.; Van Loo, G.; Danese, S.; Huth, M.; Nikolaev, A.; Neufert, C.; Madison, B.; et al. Epithelial NEMO links innate immunity to chronic intestinal inflammation. Nature 2007, 446, 557–561.
- Neurath, M.F.; Fuss, I.; Kelsall, B.L.; Stüber, E.; Strober, W. Antibodies to interleukin 12 abrogate established experimental colitis in mice. J. Exp. Med. 1995, 182, 1281–1290.
- Okayasu, I.; Hatakeyama, S.; Yamada, M.; Ohkusa, T.; Inagaki, Y.; Nakaya, R. A novel method in the induction of reliable experimental acute and chronic ulcerative colitis in mice. Gastroenterology 1990, 98, 694–702.
- Powrie, F.; Leach, M.W.; Mauze, S.; Caddie, L.B.; Coffman, R.L. Phenotypically distinct subsets of CD4+ T cells induce or protect from chronic intestinal inflammation in C. B-17 scid mice. Int. Immunol. 1993, 5, 1461–1471.
- Heller, F.; Fuss, I.J.; E Nieuwenhuis, E.; Blumberg, R.S.; Strober, W. Oxazolone Colitis, a Th2 Colitis Model Resembling Ulcerative Colitis, Is Mediated by IL-13-Producing NK-T Cells. Immunity 2002, 17, 629–638.
- Yang, J.; Zhao, J.; Nakaguchi, T.; Gregersen, H. Biomechanical changes in oxazolone-induced colitis in BALB/C mice. J. Biomech. 2009, 42, 811–817.
- Kasaian, M.T.; Page, K.M.; Fish, S.; Brennan, A.; Cook, T.A.; Moreira, K.; Zhang, M.; Jesson, M.; Marquette, K.; Agostinelli, R.; et al. Therapeutic activity of an interleukin-4/interleukin-13 dual antagonist on oxazolone-induced colitis in mice. Immunology 2014, 143, 416–427.
- Wang, X.; Ouyang, Q.; Luo, W.J. Oxazolone-induced murine model of ulcerative colitis. Chin. J. Dig. Dis. 2004, 5, 165–168.
- Strober, W.; Fuss, I.J. Proinflammatory Cytokines in the Pathogenesis of Inflammatory Bowel Diseases. Gastroenterology 2011, 140, 1756–1767.e1.
- Fuss, I.J.; Strober, W. The role of IL-13 and NK T cells in experimental and human ulcerative colitis. Mucosal Immunol. 2008, 1 (Suppl. 1), S31–S33.
- Kiesler, P.; Fuss, I.J.; Strober, W. Experimental Models of Inflammatory Bowel Diseases. Cell. Mol. Gastroenterol. Hepatol. 2015, 1, 154–170.
- Wirtz, S.; Popp, V.; Kindermann, M.; Gerlach, K.; Weigmann, B.; Fichtner-Feigl, S.; Neurath, M.F. Chemically induced mouse models of acute and chronic intestinal inflammation. Nat. Protoc. 2017, 12, 1295–1309.
- Fuss, I.J.; Heller, F.; Boirivant, M.; Leon, F.; Yoshida, M.; Fichtner-Feigl, S.; Yang, Z.; Exley, M.; Kitani, A.; Blumberg, R.S.; et al. Nonclassical CD1d-restricted NK T cells that produce IL-13 characterize an atypical Th2 response in ulcerative colitis. J. Clin. Investig. 2004, 113, 1490–1497.
- Hoving, J.C.; Cutler, A.; Leeto, M.; Horsnell, W.G.C.; Dewals, B.G.; Nieuwenhuizen, N.; Brombacher, F. Interleukin 13-mediated colitis in the absence of IL-4Rα signalling. Gut 2017, 66, 2037–2039.
- Karmele, E.P.; Pasricha, T.S.; Ramalingam, T.R.; Thompson, R.W.; Iii, R.L.G.; Knilans, K.J.; Hegen, M.; Farmer, M.; Jin, F.; Kleinman, A.; et al. Anti-IL-13Rα2 therapy promotes recovery in a murine model of inflammatory bowel disease. Mucosal Immunol. 2019, 12, 1174–1186.
- Kronenberg, M.; Gapin, L. The unconventional lifestyle of NKT cells. Nat. Rev. Immunol. 2002, 2, 557–568.
- Velde, A.A.T.; Verstege, M.I.; Hommes, D.W. Critical appraisal of the current practice in murine TNBS-induced colitis. Inflamm. Bowel Dis. 2006, 12, 995–999.
- Fichtner-Feigl, S.; Fuss, I.J.; Preiß, J.; Strober, W.; Kitani, A. Treatment of murine Th1- and Th2-mediated inflammatory bowel disease with NF- B decoy oligonucleotides. J. Clin. Investig. 2005, 115, 3057–3071.
- Fuss, I.J.; Neurath, M.; Boirivant, M.; Klein, J.S.; De La Motte, C.; A Strong, S.; Fiocchi, C.; Strober, W. Disparate CD4+ lamina propria (LP) lymphokine secretion profiles in inflammatory bowel disease. Crohn’s disease LP cells manifest increased secretion of IFN-gamma, whereas ulcerative colitis LP cells manifest increased secretion of IL-5. J. Immunol. 1996, 157, 1261–1270.
- Dohi, T.; Fujihashi, K.; Rennert, P.D.; Iwatani, K.; Kiyono, H.; McGhee, J.R. Hapten-induced Colitis Is Associated with Colonic Patch Hypertrophy and T Helper Cell 2–Type Responses. J. Exp. Med. 1999, 189, 1169–1180.
- Croxford, A.L.; Kulig, P.; Becher, B. IL-12-and IL-23 in health and disease. Cytokine Growth Factor Rev. 2014, 25, 415–421.
- Langrish, C.L.; McKenzie, B.S.; Wilson, N.J.; Malefyt, R.D.W.; Kastelein, R.A.; Cua, D.J. IL-12 and IL-23: Master regulators of innate and adaptive immunity. Immunol. Rev. 2004, 202, 96–105.
- Fuss, I.J.; Becker, C.; Yang, Z.; Groden, C.; Hornung, R.L.; Heller, F.; Neurath, M.F.; Strober, W.; Mannon, P.J. Both IL-12p70 and IL-23 are synthesized during active Crohnʼs disease and are down-regulated by treatment with anti-IL-12 p40 monoclonal antibody. Inflamm. Bowel Dis. 2006, 12, 9–15.
- Mannon, P.J.; Fuss, I.J.; Mayer, L.; Elson, C.O.; Sandborn, W.J.; Present, D.; Dolin, B.; Goodman, N.; Groden, C.; Hornung, R.L.; et al. Anti–Interleukin-12 Antibody for Active Crohn’s Disease. N. Engl. J. Med. 2004, 351, 2069–2079.
- Sands, B.E.; Sandborn, W.J.; Panaccione, R.; O’Brien, C.D.; Zhang, H.; Johanns, J.; Adedokun, O.J.; Li, K.; Peyrin-Biroulet, L.; Van Assche, G.; et al. Ustekinumab as Induction and Maintenance Therapy for Ulcerative Colitis. N. Engl. J. Med. 2019, 381, 1201–1214.
- Sewell, G.W.; Kaser, A. Interleukin-23 in the Pathogenesis of Inflammatory Bowel Disease and Implications for Therapeutic Intervention. J. Crohn’s Colitis 2022, 16, ii3–ii19.
- Laroui, H.; Ingersoll, S.A.; Liu, H.C.; Baker, M.T.; Ayyadurai, S.; Charania, M.A.; Laroui, F.; Yan, Y.; Sitaraman, S.V.; Merlin, D. Dextran Sodium Sulfate (DSS) Induces Colitis in Mice by Forming Nano-Lipocomplexes with Medium-Chain-Length Fatty Acids in the Colon. PLoS ONE 2012, 7, e32084.
- Kitajima, S.; Takuma, S.; Morimoto, M. Tissue Distribution of Dextran Sulfate Sodium(DSS) in the Acute Phase of Murine DSS-Induced Colitis. J. Vet. Med. Sci. 1999, 61, 67–70.
- Chassaing, B.; Aitken, J.D.; Malleshappa, M.; Vijay-Kumar, M. Dextran Sulfate Sodium (DSS)-Induced Colitis in Mice. Curr. Protoc. Immunol. 2014, 104, 15.25.1–15.25.14.
- Perše, M.; Cerar, A. Dextran Sodium Sulphate Colitis Mouse Model: Traps and Tricks. J. Biomed. Biotechnol. 2012, 2012, 718617.
- Dieleman, L.A.; Ridwan, B.U.; Tennyson, G.S.; Beagley, K.W.; Bucy, R.; Elson, C.O. Dextran sulfate sodium-induced colitis occurs in severe combined immunodeficient mice. Gastroenterology 1994, 107, 1643–1652.
- Kim, T.W.; Seo, J.N.; Suh, Y.H.; Park, H.J.; Kim, J.H.; Kim, J.Y.; Oh, K.I. Involvement of lymphocytes in dextran sulfate sodium-induced experimental colitis. World J. Gastroenterol. 2006, 12, 302–305.
- Tanaka, M.; Riddell, R.H.; Saito, H.; Soma, Y.; Hidaka, H.; Kudo, H. Morphologic Criteria Applicable to Biopsy Specimens for Effective Distinction of Inflammatory Bowel Disease from Other Forms of Colitis and of Crohn’s Disease from Ulcerative Colitis. Scand. J. Gastroenterol. 1999, 34, 55–67.
- Stevceva, L.; Pavli, P.; Husband, A.; Ramsay, A.; Doe, W. Dextran sulphate sodium-induced colitis is ameliorated in interleukin 4 deficient mice. Genes Immun. 2001, 2, 309–316.
- Fujino, S.; Andoh, A.; Bamba, S.; Ogawa, A.; Hata, K.; Araki, Y.; Bamba, T.; Fujiyama, Y. Increased expression of interleukin 17 in inflammatory bowel disease. Gut 2003, 52, 65–70.
- Gheita, T.A.; El Gazzar, I.I.; El-Fishawy, H.S.; Aboul-Ezz, M.A.; Kenawy, S.A. Involvement of IL-23 in enteropathic arthritis patients with inflammatory bowel disease: Preliminary results. Clin. Rheumatol. 2014, 33, 713–717.
- Jiang, W.; Su, J.; Zhang, X.; Cheng, X.; Zhou, J.; Shi, R.; Zhang, H. Elevated levels of Th17 cells and Th17-related cytokines are associated with disease activity in patients with inflammatory bowel disease. Agents Actions 2014, 63, 943–950.
- Lucaciu, L.A.; Ilieș, M.; Vesa, C.; Seicean, R.; Din, S.; Iuga, C.A.; Seicean, A. Serum Interleukin (IL)-23 and IL-17 Profile in Inflammatory Bowel Disease (IBD) Patients Could Differentiate between Severe and Non-Severe Disease. J. Pers. Med. 2021, 11, 1130.
- Mirsattari, D.; Seyyedmajidi, M.; Zojaji, H.; Haghazali, M.; Orimi, P.G.; Shoushtarizadeh, T.; Almasi, S. The relation between the level of interleukin-23 with duration and severity of ulcerative colitis. Gastroenterol. Hepatol. Bed Bench 2012, 5, 49–53.
- Oppmann, B.; Lesley, R.; Blom, B.; Timans, J.C.; Xu, Y.; Hunte, B.; Vega, F.; Yu, N.; Wang, J.; Singh, K.; et al. Novel p19 Protein Engages IL-12p40 to Form a Cytokine, IL-23, with Biological Activities Similar as Well as Distinct from IL-12. Immunity 2000, 13, 715–725.
- Aggarwal, S.; Ghilardi, N.; Xie, M.-H.; de Sauvage, F.J.; Gurney, A.L. Interleukin-23 Promotes a Distinct CD4 T Cell Activation State Characterized by the Production of Interleukin-17. J. Biol. Chem. 2003, 278, 1910–1914.
- Aden, K.; Rehman, A.; Falk-Paulsen, M.; Secher, T.; Kuiper, J.; Tran, F.; Pfeuffer, S.; Sheibani-Tezerji, R.; Breuer, A.; Luzius, A.; et al. Epithelial IL-23R Signaling Licenses Protective IL-22 Responses in Intestinal Inflammation. Cell Rep. 2016, 16, 2208–2218.
- Zenewicz, L.A.; Yancopoulos, G.D.; Valenzuela, D.M.; Murphy, A.J.; Stevens, S.; Flavell, R.A. Innate and Adaptive Interleukin-22 Protects Mice from Inflammatory Bowel Disease. Immunity 2008, 29, 947–957.
- Sugimoto, K.; Ogawa, A.; Mizoguchi, E.; Shimomura, Y.; Andoh, A.; Bhan, A.K.; Blumberg, R.S.; Xavier, R.J.; Mizoguchi, A. IL-22 ameliorates intestinal inflammation in a mouse model of ulcerative colitis. J. Clin. Investig. 2008, 118, 534–544.
- Vlantis, K.; Polykratis, A.; Welz, P.-S.; van Loo, G.; Pasparakis, M.; Wullaert, A. TLR-independent anti-inflammatory function of intestinal epithelial TRAF6 signalling prevents DSS-induced colitis in mice. Gut 2015, 65, 935–943.
- Loftus, E.V., Jr. Clinical epidemiology of inflammatory bowel disease: Incidence, prevalence, and environmental influences. Gastroenterology 2004, 126, 1504–1517.
- Heyman, M.B.; Kirschner, B.S.; Gold, B.D.; Ferry, G.; Baldassano, R.; Cohen, S.A.; Winter, H.S.; Fain, P.; King, C.; Smith, T.; et al. Children with early-onset inflammatory bowel disease (IBD): Analysis of a pediatric IBD consortium registry. J. Pediatr. 2005, 146, 35–40.
- Zhu, L.; Shi, T.; Zhong, C.; Wang, Y.; Chang, M.; Liu, X. IL-10 and IL-10 Receptor Mutations in Very Early Onset Inflammatory Bowel Disease. Gastroenterol. Res. 2017, 10, 65–69.
- Berg, D.; Davidson, N.; Kühn, R.; Muller, W.; Menon, S.; Holland, G.; Thompson-Snipes, L.; Leach, M.W.; Rennick, D. Enterocolitis and colon cancer in interleukin-10-deficient mice are associated with aberrant cytokine production and CD4(+) TH1-like responses. J. Clin. Investig. 1996, 98, 1010–1020.
- Shouval, D.S.; Biswas, A.; Goettel, J.A.; McCann, K.; Conaway, E.; Redhu, N.S.; Mascanfroni, I.D.; Al Adham, Z.; Lavoie, S.; Ibourk, M.; et al. Interleukin-10 Receptor Signaling in Innate Immune Cells Regulates Mucosal Immune Tolerance and Anti-Inflammatory Macrophage Function. Immunity 2014, 40, 706–719.
- Spencer, S.D.; Di Marco, F.; Hooley, J.; Pitts-Meek, S.; Bauer, M.; Ryan, A.M.; Sordat, B.; Gibbs, V.C.; Aguet, M. The Orphan Receptor CRF2-4 Is an Essential Subunit of the Interleukin 10 Receptor. J. Exp. Med. 1998, 187, 571–578.
- Mendoza, J.L.; Schneider, W.M.; Hoffmann, H.-H.; Vercauteren, K.; Jude, K.M.; Xiong, A.; Moraga, I.; Horton, T.M.; Glenn, J.S.; de Jong, Y.P.; et al. The IFN-λ-IFN-λR1-IL-10Rβ Complex Reveals Structural Features Underlying Type III IFN Functional Plasticity. Immunity 2017, 46, 379–392.
- Sellon, R.K.; Tonkonogy, S.; Schultz, M.; Dieleman, L.A.; Grenther, W.; Balish, E.; Rennick, D.M.; Sartor, R.B. Resident Enteric Bacteria Are Necessary for Development of Spontaneous Colitis and Immune System Activation in Interleukin-10-Deficient Mice. Infect. Immun. 1998, 66, 5224–5231.
- Colombel, J.-F.; Lémann, M.; Cassagnou, M.; Bouhnik, Y.; Duclos, B.; Dupas, J.-L.; Notteghem, B.; Mary, J.-Y. A Controlled Trial Comparing Ciprofloxacin With Mesalazine for The Treatment of Active Crohn’s Disease. Am. J. Gastroenterol. 1999, 94, 674–678.
- Sutherland, L.; Singleton, J.; Sessions, J.; Hanauer, S.; Krawitt, E.; Rankin, G.; Summers, R.; Mekhjian, H.; Greenberger, N.; Kelly, M. Double blind, placebo controlled trial of metronidazole in Crohn’s disease. Gut 1991, 32, 1071–1075.
- Madsen, K.L.; Doyle, J.S.; Tavernini, M.M.; Jewell, L.D.; Rennie, R.P.; Fedorak, R.N. Antibiotic therapy attenuates colitis in interleukin 10 gene–deficient mice. Gastroenterology 2000, 118, 1094–1105.
- Gunasekera, D.C.; Ma, J.; Vacharathit, V.; Shah, P.; Ramakrishnan, A.; Uprety, P.; Shen, Z.; Sheh, A.; Brayton, C.F.; Whary, M.T.; et al. The development of colitis in Il10−/− mice is dependent on IL-22. Mucosal Immunol. 2020, 13, 493–506.
- Ott, S.J.; Musfeldt, M.; Wenderoth, D.F.; Hampe, J.; Brant, O.; Fölsch, U.R.; Timmis, K.N.; Schreiber, S. Reduction in diversity of the colonic mucosa associated bacterial microflora in patients with active inflammatory bowel disease. Gut 2004, 53, 685–693.
- Aranda, R.; Sydora, B.C.; McAllister, P.L.; Binder, S.W.; Yang, H.Y.; Targan, S.R.; Kronenberg, M. Analysis of intestinal lymphocytes in mouse colitis mediated by transfer of CD4+, CD45RBhigh T cells to SCID recipients. J. Immunol. 1997, 158, 3464–3473.
- Feng, T.; Wang, L.; Schoeb, T.R.; Elson, C.O.; Cong, Y. Microbiota innate stimulation is a prerequisite for T cell spontaneous proliferation and induction of experimental colitis. J. Exp. Med. 2010, 207, 1321–1332.
- Mottet, C.; Uhlig, H.H.; Powrie, F. Cutting Edge: Cure of Colitis by CD4+CD25+ Regulatory T Cells. J. Immunol. 2003, 170, 3939–3943.
- Kjellev, S.; Lundsgaard, D.; Poulsen, S.S.; Markholst, H. Reconstitution of Scid mice with CD4+CD25− T cells leads to rapid colitis: An improved model for pharmacologic testing. Int. Immunopharmacol. 2006, 6, 1341–1354.
- Powrie, F.; Leach, M.W.; Mauze, S.; Menon, S.; Caddle, L.B.; Coffman, R.L. Inhibition of Thl responses prevents inflammatory bowel disease in scid mice reconstituted with CD45RBhi CD4+ T cells. Immunity 1994, 1, 553–562.
- Ahern, P.P.; Schiering, C.; Buonocore, S.; McGeachy, M.J.; Cua, D.J.; Maloy, K.J.; Powrie, F. Interleukin-23 Drives Intestinal Inflammation through Direct Activity on T Cells. Immunity 2010, 33, 279–288.
- Hue, S.; Ahern, P.; Buonocore, S.; Kullberg, M.C.; Cua, D.J.; McKenzie, B.S.; Powrie, F.; Maloy, K.J. Interleukin-23 drives innate and T cell–mediated intestinal inflammation. J. Exp. Med. 2006, 203, 2473–2483.
- Brasseit, J.; Chung, C.K.C.K.; Noti, M.; Zysset, D.; Hoheisel-Dickgreber, N.; Genitsch, V.; Corazza, N.; Mueller, C. Divergent Roles of Interferon-γ and Innate Lymphoid Cells in Innate and Adaptive Immune Cell-Mediated Intestinal Inflammation. Front. Immunol. 2018, 9, 23.
- Harbour, S.N.; Maynard, C.L.; Zindl, C.L.; Schoeb, T.R.; Weaver, C.T. Th17 cells give rise to Th1 cells that are required for the pathogenesis of colitis. Proc. Natl. Acad. Sci. USA 2015, 112, 7061–7066.
- Poholek, C.; Dulson, S.J.; Zajac, A.J.; Harrington, L.E. IL-21 Controls ILC3 Cytokine Production and Promotes a Protective Phenotype in a Mouse Model of Colitis. ImmunoHorizons 2019, 3, 194–202.
- Bosma, G.C.; Fried, M.; Custer, R.P.; Carroll, A.; Gibson, D.M.; Bosma, M.J. Evidence of functional lymphocytes in some (leaky) scid mice. J. Exp. Med. 1988, 167, 1016–1033.
- Karo, J.M.; Schatz, D.G.; Sun, J.C. The RAG Recombinase Dictates Functional Heterogeneity and Cellular Fitness in Natural Killer Cells. Cell 2014, 159, 94–107.
- Kwan, A.; Abraham, R.S.; Currier, R.; Brower, A.; Andruszewski, K.; Abbott, J.; Baker, M.; Ballow, M.; Bartoshesky, L.E.; Bonilla, F.A.; et al. Newborn Screening for Severe Combined Immunodeficiency in 11 Screening Programs in the United States. JAMA 2014, 312, 729–738.


