Crohn’s disease (CD) and ulcerative colitis (UC) are both highly inflammatory diseases of the gastrointestinal tract, collectively known as inflammatory bowel disease (IBD). Although the cause of IBD is still unclear, several experimental IBD murine models have enabled researchers to make great inroads into understanding human IBD pathology. Murine experimental models of human IBD exhibit immune pathological signatures resembling Crohn’s disease (CD) or ulcerative colitis (UC). These models include the chemical-induced trinitrobenzene sulfonic acid (TNBS) model, oxazolone and dextran sulfate sodium (DSS) models, the gene-deficient I-kappa-B kinase gamma (Iκκ-γ) and interleukin(IL)-10 models, and the CD4+ T-cell transfer model [4,5,6,7,8,9]. Although most pre-clinical murine models do not fully recapitulate the complexity of human IBD, these models have added to our knowledge about the causes of disease and have provided targets for developing new treatments.
- inflammatory bowel disease
- ulcerative colitis
- Crohn’s disease
- murine models
1. Introduction
2. Chemical Induced Colitis
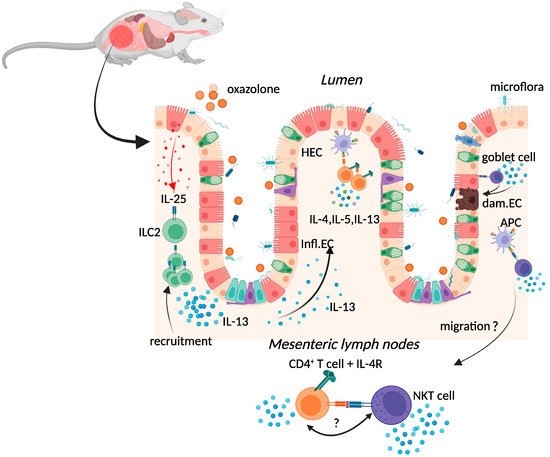
2.1. Oxazolone Colitis
2.2. TNBS-Induced Colitis
This pre-clinical model also utilises intrarectal administration of a haptenising agent in ethanol: 2,4,6-trinitrobenzene sulfonic acid (TNBS). Administration of 0.5 mg of TNBS in 50% ethanol to mice resulted in chronic transmural colitis, characterised by diarrhoea, weight loss, and rectal prolapse, pathology that mimics some characteristics of CD in humans [4][7]. Although one administration of TNBS resulted in acute chemical damage to the gut epithelium, inflammation was self-limiting, rather than the chronic inflammation seen in human disease [4][7]. Differing responses in mice are apparent in this acute model of colitis, varying according to several factors including age, genetic background, and TNBS dose [19][31]. To achieve chronic colitis, this model was developed by pre-sensitising the skin with 1% TNBS, followed by up to six repeated weekly intrarectal administrations of increasing doses of TNBS [14][17]. This model resembled the chronic phase of CD and was accompanied by production of IL-23 and IL-17 by lamina propria cells [20][32]. Isolated lamina propria CD4+ T cells from mice given TNBS secreted high levels of the Th1 cytokine interferon (IFN)-γ, resembling the cytokine profile produced by isolated lamina propria CD4+ T cells from CD patients [21][33]. This distinguished them from the Th2 profile of the same cells isolated from UC patients, or mice given oxazolone [21][33]. Although antibodies against IL-12, a pivotal cytokine for Th1 differentiation, abrogated established colitis and the initiation of TNBS-disease in BALB/c mice [4][7], TNBS-dependent colitis was exacerbated in IFN-γ-deficient mice on a BALB/c background [22][34]. The clinical importance of the TNBS model is demonstrated by the translation of Neurath’s anti-IL-12 antibody findings from TNBS murine experiments to successful human trials [23][35]. Importantly, the antibody used in these same studies was later found to react with the promiscuous p40 subunit shared by both IL-12 (a 70 kDa heterodimer of the p40 and a p35 subunit) and IL-23 (heterodimer of the p40 and a p19 subunit) (summarizeviewed in [24][36]). Clinical trials with “brakinumab”, a monoclonal antibody recognising the human p40 subunit, downregulated both IL-12p70 and IL-23 secretion [25][37] and resulted in a clinical improvement in patients with active CD [26][38]. Drug development of brakinumab was discontinued due to the existence of another IL-12/IL-23 inhibitor, Stelera (ustekinumab), on the market, which significantly increased the induction and maintenance of clinical remission in patients with UC [27][39]. Despite the findings of exacerbated TNBS-colitis in mice lacking the p19 subunit of IL-23, novel therapies targeting IL-23 and the IL-23R have been developed and deemed successful when tested in clinical trials of patients with IBD [28][40].2.3. Dextran-Sulphate-Sodium-Induced Colitis
Dextran sulphate sodium (DSS) colitis is the most widely used experimental murine model of colitis, established by Okayasu in 1990 through the administration of DSS with a molecular weight of 40–50 kDa in drinking water [5][8]. DSS is thought to form nano-lipid vesicles with medium-chain fatty acids (MCFAs) in the colon, which fuse with colonocyte membranes and increase inflammatory cytokine levels [29][54]. A high-fat diet rich in MCFAs exacerbated weight loss, inflammatory cytokine expression, and colon shortening in this model, with dodecanoic acid favouring disruption of intestinal barrier function and increased vesicle formation in vitro [29][54]. One day after administration, DSS particles were present systemically in Kupffer cells of the liver, in macrophages of the mesenteric lymph node, and in the lamina propria of the large intestine [30][55]. DSS administration was also characterised by erosion of the intestinal epithelium, inflammatory infiltration of the large intestine, and dysbiosis of the intestinal microbiome [5][8]. While these features are similar to those found in human disease, the transmural inflammation apparent in TNBS-colitis is absent in this model [31][56]. Although repeated rounds of DSS can be administered to provide the pattern of remitting, relapsing inflammation in human IBD, some of the limitations of this model include inter-batch variability of DSS and the need to optimise DSS dose, given the impact of the intestinal microbiome on disease [31][32][56,57].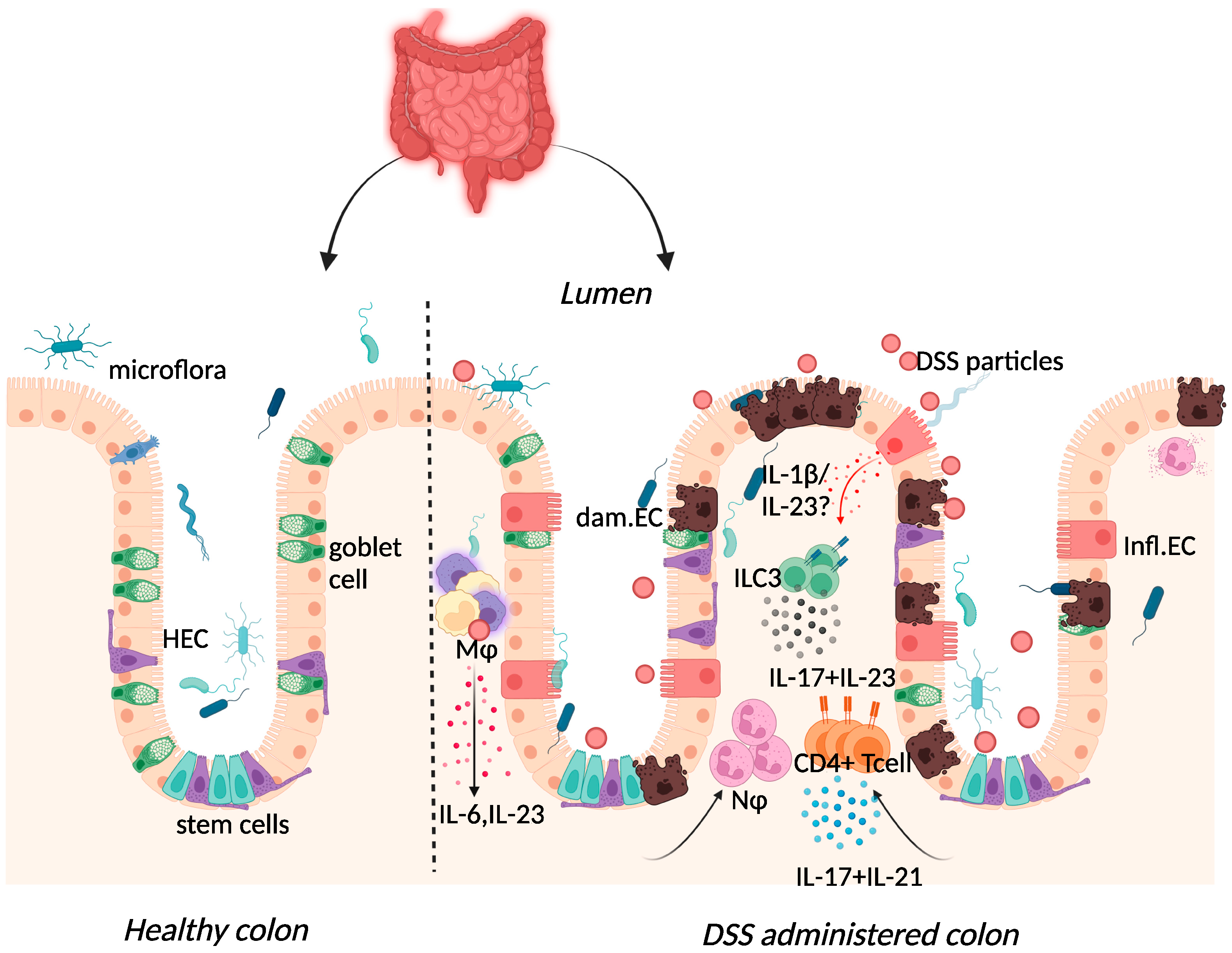
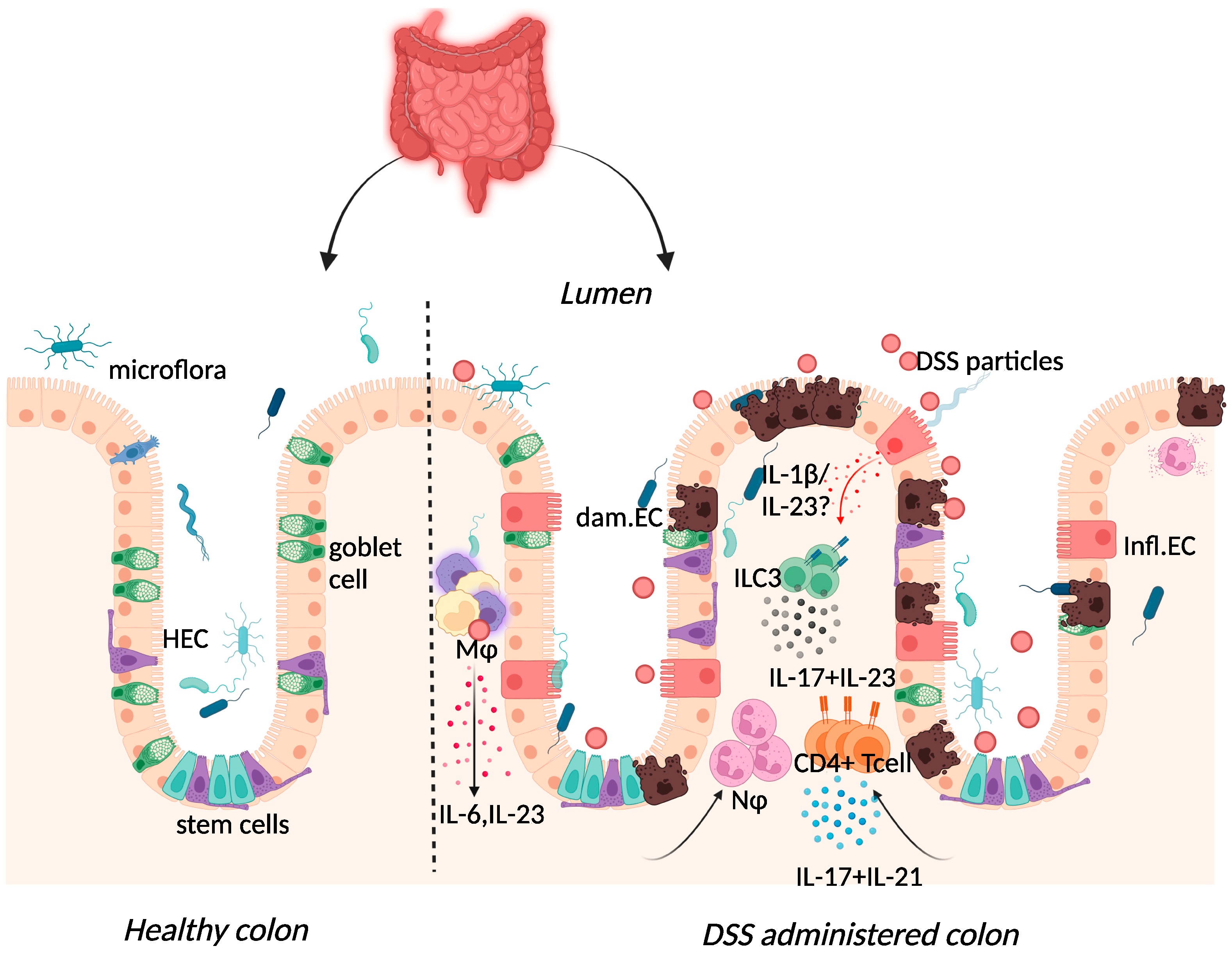 Figure 2. Immune response to DSS administration. DSS administration results in epithelial release of IL-1β, activation of ILC3, and release of IL-23. IL-23 release results in the influx of neutrophils and CD4+ T cells, which further respond through enhanced IL-17 signalling. The resulting chronic inflammatory responses result in goblet cell depletion, increased intestinal permeability, and increased adhesion of commensal intestinal microbiota to the epithelium. Key: Inflamed epithelial cells (Infl.EC), healthy epithelial cells (HEC), damaged epithelial cell (dam.EC), macrophages (MΦ), neutrophils (NΦ). Created with BioRender.com, accessed on 18 August 2022.
DSS-colitis can be induced in immunodeficient mice including recombination-activating gene (RAG)-1-deficient and severe combined immune deficient (SCID) mice, suggesting the dispensability of the adaptive immune system in initiating disease [33][34][62,63]. Although Kim et al. demonstrated colitis induction in RAG-1-deficient mice, the resultant mild colitis in these mice compared to their wild-type counterparts insinuates that lymphocytes may be necessary for subsequent colitis progression [34][63]. Histological assessment of biopsy specimens from IBD patients correlated UC and CD with severe mononuclear cell infiltration and basal plasmacytosis (plasma B cells) [35][64]. In the acute phase of the DSS-colitis model, this infiltrate consisted of innate macrophage, neutrophil, and eosinophil populations recruited following increased cytokine and chemokine expression [36][60].
Elevated expression of IL-17 and IL-23 was reported in IBD patients and in DSS-colitis, where expression of the two cytokines was intertwined (Figure 2) [25][37][38][39][40][41][37,65,66,67,68,69]. The use of the DSS-colitis model to test the role of these cytokines in disease has revolutionised not only the possible interventions available for patients but has also developed our understanding of mucosal immunology. At steady state, the p19 subunit of IL-23 is highly expressed within Peyer’s patches and the thymus, as well as in polarised Th1 cells, activated macrophages, and dendritic cell populations derived from peripheral blood [42][70]. IL-23 induced proliferation of memory T cells and elevated secretion of IL-17 in vitro [43][71]. Subsequently, IL-23 signalling within intestinal epithelial cells was found to play an important role in protection against DSS colitis by regulating regenerating-islet-derived protein 3-beta (Reg3β)-dependent control of flagellated intestinal bacterial abundance and promoting IL-22 production [44][72]. Indeed DSS-colitis was exacerbated in IL-22-deficient mice [45][73] and blockade of IL-22 expression delayed recovery from DSS-colitis and exacerbated disease scores [46][74].
Figure 2. Immune response to DSS administration. DSS administration results in epithelial release of IL-1β, activation of ILC3, and release of IL-23. IL-23 release results in the influx of neutrophils and CD4+ T cells, which further respond through enhanced IL-17 signalling. The resulting chronic inflammatory responses result in goblet cell depletion, increased intestinal permeability, and increased adhesion of commensal intestinal microbiota to the epithelium. Key: Inflamed epithelial cells (Infl.EC), healthy epithelial cells (HEC), damaged epithelial cell (dam.EC), macrophages (MΦ), neutrophils (NΦ). Created with BioRender.com, accessed on 18 August 2022.
DSS-colitis can be induced in immunodeficient mice including recombination-activating gene (RAG)-1-deficient and severe combined immune deficient (SCID) mice, suggesting the dispensability of the adaptive immune system in initiating disease [33][34][62,63]. Although Kim et al. demonstrated colitis induction in RAG-1-deficient mice, the resultant mild colitis in these mice compared to their wild-type counterparts insinuates that lymphocytes may be necessary for subsequent colitis progression [34][63]. Histological assessment of biopsy specimens from IBD patients correlated UC and CD with severe mononuclear cell infiltration and basal plasmacytosis (plasma B cells) [35][64]. In the acute phase of the DSS-colitis model, this infiltrate consisted of innate macrophage, neutrophil, and eosinophil populations recruited following increased cytokine and chemokine expression [36][60].
Elevated expression of IL-17 and IL-23 was reported in IBD patients and in DSS-colitis, where expression of the two cytokines was intertwined (Figure 2) [25][37][38][39][40][41][37,65,66,67,68,69]. The use of the DSS-colitis model to test the role of these cytokines in disease has revolutionised not only the possible interventions available for patients but has also developed our understanding of mucosal immunology. At steady state, the p19 subunit of IL-23 is highly expressed within Peyer’s patches and the thymus, as well as in polarised Th1 cells, activated macrophages, and dendritic cell populations derived from peripheral blood [42][70]. IL-23 induced proliferation of memory T cells and elevated secretion of IL-17 in vitro [43][71]. Subsequently, IL-23 signalling within intestinal epithelial cells was found to play an important role in protection against DSS colitis by regulating regenerating-islet-derived protein 3-beta (Reg3β)-dependent control of flagellated intestinal bacterial abundance and promoting IL-22 production [44][72]. Indeed DSS-colitis was exacerbated in IL-22-deficient mice [45][73] and blockade of IL-22 expression delayed recovery from DSS-colitis and exacerbated disease scores [46][74].
3. Spontaneous Colitis
3.1. Iκκ-γ (NEMO) Deficiency Colitis
Conditional ablation of the NF-kB essential modulator (NEMO), also known as I-kappa-B kinase gamma (Iκκ-γ), within the intestinal epithelium, resulted in spontaneous colitis in mice [3][6]. Chronic disease in intestinal-epithelial-cell-specific NEMO-deficient mice was associated with TNFR1-dependent colonic epithelial cell death, compromised epithelial integrity, bacterial translocation in the colon, immune cell infiltration, and increased expression of pro-inflammatory cytokines including TNF-α [3][6]. Absence of disease in double-deficient (NEMOIEC−KO + MYD88−/−) mice, lacking NEMO and the important bacterial sensor myeloid differentiation primary response 88 (MYD88), supported a role for the gut microbiota in driving colitis [3][6]. Indeed NEMOIEC−KO mice raised under germ-free conditions did not develop spontaneous colitis, whereas co-housing of these mice with specific pathogen-free animals restored disease [47][97].3.2. Interleukin-10 (IL-10) Deficiency Colitis
Although IBD is common among adults, childhood IBD constitutes about a quarter of all patients with IBD [48][101]. Approximately 15% of childhood IBD occurs in children <6 years old and is termed very early onset IBD (VEO-IBD) [49][102]. While most childhood IBD cases are polygenic in nature, many children with VEO-IBD have an underlying monogenetic disorder that results in severe enterocolitis, including mutations in IL-10 and/or IL-10R (summarizeviewed in [50][103]). Mice deficient in IL-10 develop spontaneous enterocolitis, characterised by progressive cellular infiltration of the cecum, colon, rectum, and small intestine, with transmural lesions and a high incidence of colorectal adenocarcinomas observed in 6-month-old mice [2][51][5,104]. Mice lacking IL-10 receptor β also developed spontaneous enterocolitis [52][53][105,106]. However, this receptor is shared with other type II cytokine receptors including those specific for IL-22, IL-26, and lambda interferons, in addition to IL-10 [54][107]. An important role for the gut microbiota in influencing the general enterocolitis seen in IL-10-deficient mice was implicated by a lack of disease in mice housed in specific pathogen-free conditions, or in germ-free conditions [2][55][5,112]. Treatment of IL-10-deficient mice with two different antibiotics, both shown to improve scores in patients with Crohn’s disease [56][57][113,114], attenuated the development of spontaneous colitis [58][115]. Similar to IBD patients, colitic IL-10-deficient mice exhibited a markedly reduced species diversity in their faecal microbiome when compared to disease-free controls [55][59][60][111,112,116]. In addition, IL-10/IL-22 double-deficient mice lacking colitis exhibited higher microbial diversity when compared to IL-10-deficient mice [59][111].4. Immune Cell Induced Colitis
T-Cell Adoptive Transfer Model
The transfer of murine CD45RBhighCD4+ T cells from healthy donors to severe combined immunodeficiency (SCID) (Figure 3) mice resulted in the development of a lethal wasting disease, an influx of inflammatory cells, and increased inflammatory cytokine production in the colon of the recipient [6][9].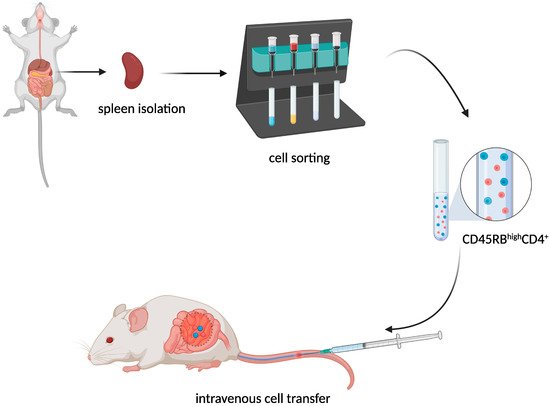
[1][2][3][4][5][6][7][8][9][10][11][12][13][14][15][16][17][18][19][20][21][22][23][24][25][26][27][28][29][30][31][32][33][34][35][36][37][38][39][40][41][42][43][44][45][46][47][48][49][50][51][52][53][54][55][56][57][58][59][60][61][62][63][64][65][66][67][68][69][70][71][72][73][74][75][76][77][78][79][80][81][82][83][84][85][86][87][88][89][90][91][92][93][94][95][96][97][98][99][100][101][102][103][104][105][106][107][108][109][110][111][112][113][114][115][116][117][118][119][120][121][122][123][124][125][126][127][128][129][130][131][132][133][134][135][136][137][138][139][140][141][142][143][144][145][146][147][148][149][150][151][152][153][154][155]
References
- Boirivant, M.; Fuss, I.J.; Chu, A.; Strober, W. Oxazolone Colitis: A Murine Model of T Helper Cell Type 2 Colitis Treatable with Antibodies to Interleukin 4. J. Exp. Med. 1998, 188, 1929–1939. Ng, S.C.; Shi, H.Y.; Hamidi, N.; Underwood, F.E.; Tang, W.; Benchimol, E.I.; Panaccione, R.; Ghosh, S.; Wu, J.C.Y.; Chan, F.K.L.; et al. Worldwide incidence and prevalence of inflammatory bowel disease in the 21st century: A systematic review of population-based studies. Lancet 2017, 390, 2769–2778. https://doi.org/10.1016/s0140-6736(17)32448-0.
- Kühn, R.; Löhler, J.; Rennick, D.; Rajewsky, K.; Müller, W. Interleukin-10-deficient mice develop chronic enterocolitis. Cell 1993, 75, 263–274. Alatab, S.; Sepanlou, S.G.; Ikuta, K.; Vahedi, H.; Bisignano, C.; Safiri, S.; Sadeghi, A.; Nixon, M.R.; Abdoli, A.; Abolhassani, H.; et al. The global, regional, and national burden of inflammatory bowel disease in 195 countries and territories, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet Gastroenterol. Hepatol. 2020, 5, 17–30. https://doi.org/10.1016/s2468-1253(19)30333-4.
- Nenci, A.; Becker, C.; Wullaert, A.; Gareus, R.; Van Loo, G.; Danese, S.; Huth, M.; Nikolaev, A.; Neufert, C.; Madison, B.; et al. Epithelial NEMO links innate immunity to chronic intestinal inflammation. Nature 2007, 446, 557–561. Kappelman, M.D.; Rifas–Shiman, S.L.; Porter, C.Q.; Ollendorf, D.A.; Sandler, R.S.; Galanko, J.A.; Finkelstein, J.A. Direct Health Care Costs of Crohn’s Disease and Ulcerative Colitis in US Children and Adults. Gastroenterology 2008, 135, 1907–1913. https://doi.org/10.1053/j.gastro.2008.09.012.
- Neurath, M.F.; Fuss, I.; Kelsall, B.L.; Stüber, E.; Strober, W. Antibodies to interleukin 12 abrogate established experimental colitis in mice. J. Exp. Med. 1995, 182, 1281–1290. Boirivant, M.; Fuss, I.J.; Chu, A.; Strober, W. Oxazolone Colitis: A Murine Model of T Helper Cell Type 2 Colitis Treatable with Antibodies to Interleukin 4. Exp. Med. 1998, 188, 1929–1939. https://doi.org/10.1084/jem.188.10.1929.
- Okayasu, I.; Hatakeyama, S.; Yamada, M.; Ohkusa, T.; Inagaki, Y.; Nakaya, R. A novel method in the induction of reliable experimental acute and chronic ulcerative colitis in mice. Gastroenterology 1990, 98, 694–702. Kühn, R.; Löhler, J.; Rennick, D.; Rajewsky, K.; Müller, W. Interleukin-10-deficient mice develop chronic enterocolitis. Cell 1993, 75, 263–274. https://doi.org/10.1016/0092-8674(93)80068-p.
- Powrie, F.; Leach, M.W.; Mauze, S.; Caddie, L.B.; Coffman, R.L. Phenotypically distinct subsets of CD4+ T cells induce or protect from chronic intestinal inflammation in C. B-17 scid mice. Int. Immunol. 1993, 5, 1461–1471. Nenci, A.; Becker, C.; Wullaert, A.; Gareus, R.; Van Loo, G.; Danese, S.; Huth, M.; Nikolaev, A.; Neufert, C.; Madison, B.; et al. Epithelial NEMO links innate immunity to chronic intestinal inflammation. Nature 2007, 446, 557–561. https://doi.org/10.1038/nature05698.
- Heller, F.; Fuss, I.J.; E Nieuwenhuis, E.; Blumberg, R.S.; Strober, W. Oxazolone Colitis, a Th2 Colitis Model Resembling Ulcerative Colitis, Is Mediated by IL-13-Producing NK-T Cells. Immunity 2002, 17, 629–638. Neurath, M.F.; Fuss, I.; Kelsall, B.L.; Stüber, E.; Strober, W. Antibodies to interleukin 12 abrogate established experimental colitis in mice. Exp. Med. 1995, 182, 1281–1290. https://doi.org/10.1084/jem.182.5.1281.
- Yang, J.; Zhao, J.; Nakaguchi, T.; Gregersen, H. Biomechanical changes in oxazolone-induced colitis in BALB/C mice. J. Biomech. 2009, 42, 811–817. Okayasu, I.; Hatakeyama, S.; Yamada, M.; Ohkusa, T.; Inagaki, Y.; Nakaya, R. A novel method in the induction of reliable experimental acute and chronic ulcerative colitis in mice. Gastroenterology 1990, 98, 694–702. https://doi.org/10.1016/0016-5085(90)90290-h.
- Kasaian, M.T.; Page, K.M.; Fish, S.; Brennan, A.; Cook, T.A.; Moreira, K.; Zhang, M.; Jesson, M.; Marquette, K.; Agostinelli, R.; et al. Therapeutic activity of an interleukin-4/interleukin-13 dual antagonist on oxazolone-induced colitis in mice. Immunology 2014, 143, 416–427. Powrie, F.; Leach, M.W.; Mauze, S.; Caddie, L.B.; Coffman, R.L. Phenotypically distinct subsets of CD4+ T cells induce or protect from chronic intestinal inflammation in C. B-17 scid Int. Immunol. 1993, 5, 1461–1471. https://doi.org/10.1093/intimm/5.11.1461.
- Wang, X.; Ouyang, Q.; Luo, W.J. Oxazolone-induced murine model of ulcerative colitis. Chin. J. Dig. Dis. 2004, 5, 165–168. Heller, F.; Fuss, I.J.; E Nieuwenhuis, E.; Blumberg, R.S.; Strober, W. Oxazolone Colitis, a Th2 Colitis Model Resembling Ulcerative Colitis, Is Mediated by IL-13-Producing NK-T Cells. Immunity 2002, 17, 629–638. https://doi.org/10.1016/s1074-7613(02)00453-3.
- Strober, W.; Fuss, I.J. Proinflammatory Cytokines in the Pathogenesis of Inflammatory Bowel Diseases. Gastroenterology 2011, 140, 1756–1767.e1. Yang, J.; Zhao, J.; Nakaguchi, T.; Gregersen, H. Biomechanical changes in oxazolone-induced colitis in BALB/C mice. Biomech. 2009, 42, 811–817. https://doi.org/10.1016/j.jbiomech.2009.01.028.
- Fuss, I.J.; Strober, W. The role of IL-13 and NK T cells in experimental and human ulcerative colitis. Mucosal Immunol. 2008, 1 (Suppl. 1), S31–S33. Kasaian, M.T.; Page, K.M.; Fish, S.; Brennan, A.; Cook, T.A.; Moreira, K.; Zhang, M.; Jesson, M.; Marquette, K.; Agostinelli, R.; et al. Therapeutic activity of an interleukin-4/interleukin-13 dual antagonist on oxazolone-induced colitis in mice. Immunology 2014, 143, 416–427. https://doi.org/10.1111/imm.12319.
- Kiesler, P.; Fuss, I.J.; Strober, W. Experimental Models of Inflammatory Bowel Diseases. Cell. Mol. Gastroenterol. Hepatol. 2015, 1, 154–170. Wang, X.; Ouyang, Q.; Luo, W.J. Oxazolone-induced murine model of ulcerative colitis. J. Dig. Dis. 2004, 5, 165–168. https://doi.org/10.1111/j.1443-9573.2004.00173.x.
- Wirtz, S.; Popp, V.; Kindermann, M.; Gerlach, K.; Weigmann, B.; Fichtner-Feigl, S.; Neurath, M.F. Chemically induced mouse models of acute and chronic intestinal inflammation. Nat. Protoc. 2017, 12, 1295–1309. Strober, W.; Fuss, I.J. Proinflammatory Cytokines in the Pathogenesis of Inflammatory Bowel Diseases. Gastroenterology 2011, 140, 1756–1767.e1. https://doi.org/10.1053/j.gastro.2011.02.016.
- Fuss, I.J.; Heller, F.; Boirivant, M.; Leon, F.; Yoshida, M.; Fichtner-Feigl, S.; Yang, Z.; Exley, M.; Kitani, A.; Blumberg, R.S.; et al. Nonclassical CD1d-restricted NK T cells that produce IL-13 characterize an atypical Th2 response in ulcerative colitis. J. Clin. Investig. 2004, 113, 1490–1497. Fuss, I.J.; Strober, W. The role of IL-13 and NK T cells in experimental and human ulcerative colitis. Mucosal Immunol. 2008, 1 (Suppl. 1), S31–S33. https://doi.org/10.1038/mi.2008.40.
- Hoving, J.C.; Cutler, A.; Leeto, M.; Horsnell, W.G.C.; Dewals, B.G.; Nieuwenhuizen, N.; Brombacher, F. Interleukin 13-mediated colitis in the absence of IL-4Rα signalling. Gut 2017, 66, 2037–2039. Kiesler, P.; Fuss, I.J.; Strober, W. Experimental Models of Inflammatory Bowel Diseases. Mol. Gastroenterol. Hepatol. 2015, 1, 154–170. https://doi.org/10.1016/j.jcmgh.2015.01.006.
- Karmele, E.P.; Pasricha, T.S.; Ramalingam, T.R.; Thompson, R.W.; Iii, R.L.G.; Knilans, K.J.; Hegen, M.; Farmer, M.; Jin, F.; Kleinman, A.; et al. Anti-IL-13Rα2 therapy promotes recovery in a murine model of inflammatory bowel disease. Mucosal Immunol. 2019, 12, 1174–1186. Wirtz, S.; Popp, V.; Kindermann, M.; Gerlach, K.; Weigmann, B.; Fichtner-Feigl, S.; Neurath, M.F. Chemically induced mouse models of acute and chronic intestinal inflammation. Protoc. 2017, 12, 1295–1309. https://doi.org/10.1038/nprot.2017.044.
- Kronenberg, M.; Gapin, L. The unconventional lifestyle of NKT cells. Nat. Rev. Immunol. 2002, 2, 557–568. Fuss, I.J.; Heller, F.; Boirivant, M.; Leon, F.; Yoshida, M.; Fichtner-Feigl, S.; Yang, Z.; Exley, M.; Kitani, A.; Blumberg, R.S.; et al. Nonclassical CD1d-restricted NK T cells that produce IL-13 characterize an atypical Th2 response in ulcerative colitis. Clin. Investig. 2004, 113, 1490–1497. https://doi.org/10.1172/jci19836.
- Velde, A.A.T.; Verstege, M.I.; Hommes, D.W. Critical appraisal of the current practice in murine TNBS-induced colitis. Inflamm. Bowel Dis. 2006, 12, 995–999. Hoving, J.C.; Cutler, A.; Leeto, M.; Horsnell, W.G.C.; Dewals, B.G.; Nieuwenhuizen, N.; Brombacher, F. Interleukin 13-mediated colitis in the absence of IL-4Rα signalling. Gut 2017, 66, 2037–2039. https://doi.org/10.1136/gutjnl-2016-313208.
- Fichtner-Feigl, S.; Fuss, I.J.; Preiß, J.; Strober, W.; Kitani, A. Treatment of murine Th1- and Th2-mediated inflammatory bowel disease with NF- B decoy oligonucleotides. J. Clin. Investig. 2005, 115, 3057–3071. Karmele, E.P.; Pasricha, T.S.; Ramalingam, T.R.; Thompson, R.W.; Iii, R.L.G.; Knilans, K.J.; Hegen, M.; Farmer, M.; Jin, F.; Kleinman, A.; et al. Anti-IL-13Rα2 therapy promotes recovery in a murine model of inflammatory bowel disease. Mucosal Immunol. 2019, 12, 1174–1186. https://doi.org/10.1038/s41385-019-0189-6.
- Fuss, I.J.; Neurath, M.; Boirivant, M.; Klein, J.S.; De La Motte, C.; A Strong, S.; Fiocchi, C.; Strober, W. Disparate CD4+ lamina propria (LP) lymphokine secretion profiles in inflammatory bowel disease. Crohn’s disease LP cells manifest increased secretion of IFN-gamma, whereas ulcerative colitis LP cells manifest increased secretion of IL-5. J. Immunol. 1996, 157, 1261–1270. Fichtner-Feigl, S.; Strober, W.; Kawakami, K.; Puri, R.K.; Kitani, A. IL-13 signaling through the IL-13α2 receptor is involved in induction of TGF-β1 production and fibrosis. Med. 2006, 12, 99–106. https://doi.org/10.1038/nm1332.
- Dohi, T.; Fujihashi, K.; Rennert, P.D.; Iwatani, K.; Kiyono, H.; McGhee, J.R. Hapten-induced Colitis Is Associated with Colonic Patch Hypertrophy and T Helper Cell 2–Type Responses. J. Exp. Med. 1999, 189, 1169–1180. Palamides, P.; Jodeleit, H.; Föhlinger, M.; Beigel, F.; Herbach, N.; Mueller, T.; Wolf, E.; Siebeck, M.; Gropp, R. A mouse model for ulcerative colitis based on NOD-scid IL2R gammanull mice reconstituted with peripheral blood mononuclear cells from affected individuals. Model. Mech. 2016, 9, 985–997. https://doi.org/10.1242/dmm.025452.
- Croxford, A.L.; Kulig, P.; Becher, B. IL-12-and IL-23 in health and disease. Cytokine Growth Factor Rev. 2014, 25, 415–421. Reinisch, W.; Panes, J.; Khurana, S.; Toth, G.; Hua, F.; Comer, G.M.; Hinz, M.; Page, K.; O’Toole, M.; Moorehead, T.M.; et al. Anrukinzumab, an anti-interleukin 13 monoclonal antibody, in active UC: Efficacy and safety from a phase IIa randomised multicentre study. Gut 2015, 64, 894–900. https://doi.org/10.1136/gutjnl-2014-308337.
- Langrish, C.L.; McKenzie, B.S.; Wilson, N.J.; Malefyt, R.D.W.; Kastelein, R.A.; Cua, D.J. IL-12 and IL-23: Master regulators of innate and adaptive immunity. Immunol. Rev. 2004, 202, 96–105. Danese, S.; Rudziński, J.; Brandt, W.; Dupas, J.-L.; Peyrin-Biroulet, L.; Bouhnik, Y.; Kleczkowski, D.; Uebel, P.; Lukas, M.; Knutsson, M.; et al. Tralokinumab for moderate-to-severe UC: A randomised, double-blind, placebo-controlled, phase IIa study. Gut 2015, 64, 243–249. https://doi.org/10.1136/gutjnl-2014-308004.
- Fuss, I.J.; Becker, C.; Yang, Z.; Groden, C.; Hornung, R.L.; Heller, F.; Neurath, M.F.; Strober, W.; Mannon, P.J. Both IL-12p70 and IL-23 are synthesized during active Crohnʼs disease and are down-regulated by treatment with anti-IL-12 p40 monoclonal antibody. Inflamm. Bowel Dis. 2006, 12, 9–15. Kronenberg, M.; Gapin, L. The unconventional lifestyle of NKT cells. Rev. Immunol. 2002, 2, 557–568. https://doi.org/10.1038/nri854.
- Mannon, P.J.; Fuss, I.J.; Mayer, L.; Elson, C.O.; Sandborn, W.J.; Present, D.; Dolin, B.; Goodman, N.; Groden, C.; Hornung, R.L.; et al. Anti–Interleukin-12 Antibody for Active Crohn’s Disease. N. Engl. J. Med. 2004, 351, 2069–2079. Hoving, J.C.; Kirstein, F.; Nieuwenhuizen, N.; Fick, L.C.; Hobeika, E.; Reth, M.; Brombacher, F. B Cells That Produce Immunoglobulin E Mediate Colitis in BALB/c Mice. Gastroenterology 2012, 142, 96–108. https://doi.org/10.1053/j.gastro.2011.09.044.
- Sands, B.E.; Sandborn, W.J.; Panaccione, R.; O’Brien, C.D.; Zhang, H.; Johanns, J.; Adedokun, O.J.; Li, K.; Peyrin-Biroulet, L.; Van Assche, G.; et al. Ustekinumab as Induction and Maintenance Therapy for Ulcerative Colitis. N. Engl. J. Med. 2019, 381, 1201–1214. Camelo, A.; Barlow, J.L.; Drynan, L.F.; Neill, D.R.; Ballantyne, S.J.; Wong, S.H.; Pannell, R.; Gao, W.; Wrigley, K.; Sprenkle, J.; et al. Blocking IL-25 signalling protects against gut inflammation in a type-2 model of colitis by suppressing nuocyte and NKT derived IL-13. Gastroenterol. 2012, 47, 1198–1211. https://doi.org/10.1007/s00535-012-0591-2.
- Sewell, G.W.; Kaser, A. Interleukin-23 in the Pathogenesis of Inflammatory Bowel Disease and Implications for Therapeutic Intervention. J. Crohn’s Colitis 2022, 16, ii3–ii19. Forkel, M.; Van Tol, S.; Höög, C.; Michaëlsson, J.; Almer, S.; Mjösberg, J. Distinct Alterations in the Composition of Mucosal Innate Lymphoid Cells in Newly Diagnosed and Established Crohn’s Disease and Ulcerative Colitis. Crohn’s Colitis 2019, 13, 67–78. https://doi.org/10.1093/ecco-jcc/jjy119.
- Laroui, H.; Ingersoll, S.A.; Liu, H.C.; Baker, M.T.; Ayyadurai, S.; Charania, M.A.; Laroui, F.; Yan, Y.; Sitaraman, S.V.; Merlin, D. Dextran Sodium Sulfate (DSS) Induces Colitis in Mice by Forming Nano-Lipocomplexes with Medium-Chain-Length Fatty Acids in the Colon. PLoS ONE 2012, 7, e32084. De Salvo, C.; Buela, K.-A.; Creyns, B.; Corridoni, D.; Rana, N.; Wargo, H.L.; Cominelli, C.L.; Delaney, P.G.; Rodriguez-Palacios, A.; Cominelli, F.; et al. NOD2 drives early IL-33–dependent expansion of group 2 innate lymphoid cells during Crohn’s disease–like ileitis. Clin. Investig. 2021, 131, . https://doi.org/10.1172/jci140624.
- Kitajima, S.; Takuma, S.; Morimoto, M. Tissue Distribution of Dextran Sulfate Sodium(DSS) in the Acute Phase of Murine DSS-Induced Colitis. J. Vet. Med. Sci. 1999, 61, 67–70. Nolte, T.; Zadeh-Khorasani, M.; Safarov, O.; Rueff, F.; Gülberg, V.; Herbach, N.; Wollenberg, A.; Mueller, T.; Siebeck, M.; Wolf, E.; et al. Oxazolone and ethanol induce colitis in non-obese diabetic-severe combined immunodeficiency interleukin-2Rγnull mice engrafted with human peripheral blood mononuclear cells. Exp. Immunol. 2013, 172, 349–362. https://doi.org/10.1111/cei.12057.
- Chassaing, B.; Aitken, J.D.; Malleshappa, M.; Vijay-Kumar, M. Dextran Sulfate Sodium (DSS)-Induced Colitis in Mice. Curr. Protoc. Immunol. 2014, 104, 15.25.1–15.25.14. Velde, A.A.T.; Verstege, M.I.; Hommes, D.W. Critical appraisal of the current practice in murine TNBS-induced colitis. Bowel Dis. 2006, 12, 995–999. https://doi.org/10.1097/01.mib.0000227817.54969.5e.
- Perše, M.; Cerar, A. Dextran Sodium Sulphate Colitis Mouse Model: Traps and Tricks. J. Biomed. Biotechnol. 2012, 2012, 718617. Fichtner-Feigl, S.; Fuss, I.J.; Preiß, J.; Strober, W.; Kitani, A. Treatment of murine Th1- and Th2-mediated inflammatory bowel disease with NF- B decoy oligonucleotides. Clin. Investig. 2005, 115, 3057–3071. https://doi.org/10.1172/jci24792.
- Dieleman, L.A.; Ridwan, B.U.; Tennyson, G.S.; Beagley, K.W.; Bucy, R.; Elson, C.O. Dextran sulfate sodium-induced colitis occurs in severe combined immunodeficient mice. Gastroenterology 1994, 107, 1643–1652. Fuss, I.J.; Neurath, M.; Boirivant, M.; Klein, J.S.; De La Motte, C.; A Strong, S.; Fiocchi, C.; Strober, W. Disparate CD4+ lamina propria (LP) lymphokine secretion profiles in inflammatory bowel disease. Crohn’s disease LP cells manifest increased secretion of IFN-gamma, whereas ulcerative colitis LP cells manifest increased secretion of IL-5. Immunol. 1996, 157, 1261–1270.
- Kim, T.W.; Seo, J.N.; Suh, Y.H.; Park, H.J.; Kim, J.H.; Kim, J.Y.; Oh, K.I. Involvement of lymphocytes in dextran sulfate sodium-induced experimental colitis. World J. Gastroenterol. 2006, 12, 302–305. Dohi, T.; Fujihashi, K.; Rennert, P.D.; Iwatani, K.; Kiyono, H.; McGhee, J.R. Hapten-induced Colitis Is Associated with Colonic Patch Hypertrophy and T Helper Cell 2–Type Responses. Exp. Med. 1999, 189, 1169–1180. https://doi.org/10.1084/jem.189.8.1169.
- Tanaka, M.; Riddell, R.H.; Saito, H.; Soma, Y.; Hidaka, H.; Kudo, H. Morphologic Criteria Applicable to Biopsy Specimens for Effective Distinction of Inflammatory Bowel Disease from Other Forms of Colitis and of Crohn’s Disease from Ulcerative Colitis. Scand. J. Gastroenterol. 1999, 34, 55–67. Croxford, A.L.; Kulig, P.; Becher, B. IL-12-and IL-23 in health and disease. Cytokine Growth Factor Rev. 2014, 25, 415–421. https://doi.org/10.1016/j.cytogfr.2014.07.017.
- Stevceva, L.; Pavli, P.; Husband, A.; Ramsay, A.; Doe, W. Dextran sulphate sodium-induced colitis is ameliorated in interleukin 4 deficient mice. Genes Immun. 2001, 2, 309–316. Langrish, C.L.; McKenzie, B.S.; Wilson, N.J.; Malefyt, R.D.W.; Kastelein, R.A.; Cua, D.J. IL-12 and IL-23: Master regulators of innate and adaptive immunity. Rev. 2004, 202, 96–105. https://doi.org/10.1111/j.0105-2896.2004.00214.x.
- Fujino, S.; Andoh, A.; Bamba, S.; Ogawa, A.; Hata, K.; Araki, Y.; Bamba, T.; Fujiyama, Y. Increased expression of interleukin 17 in inflammatory bowel disease. Gut 2003, 52, 65–70. Fuss, I.J.; Becker, C.; Yang, Z.; Groden, C.; Hornung, R.L.; Heller, F.; Neurath, M.F.; Strober, W.; Mannon, P.J. Both IL-12p70 and IL-23 are synthesized during active Crohnʼs disease and are down-regulated by treatment with anti-IL-12 p40 monoclonal antibody. Bowel Dis. 2006, 12, 9–15. https://doi.org/10.1097/01.mib.0000194183.92671.b6.
- Gheita, T.A.; El Gazzar, I.I.; El-Fishawy, H.S.; Aboul-Ezz, M.A.; Kenawy, S.A. Involvement of IL-23 in enteropathic arthritis patients with inflammatory bowel disease: Preliminary results. Clin. Rheumatol. 2014, 33, 713–717. Mannon, P.J.; Fuss, I.J.; Mayer, L.; Elson, C.O.; Sandborn, W.J.; Present, D.; Dolin, B.; Goodman, N.; Groden, C.; Hornung, R.L.; et al. Anti–Interleukin-12 Antibody for Active Crohn’s Disease. New Engl. J. Med. 2004, 351, 2069–2079. https://doi.org/10.1056/nejmoa033402.
- Jiang, W.; Su, J.; Zhang, X.; Cheng, X.; Zhou, J.; Shi, R.; Zhang, H. Elevated levels of Th17 cells and Th17-related cytokines are associated with disease activity in patients with inflammatory bowel disease. Agents Actions 2014, 63, 943–950. Sands, B.E.; Sandborn, W.J.; Panaccione, R.; O’Brien, C.D.; Zhang, H.; Johanns, J.; Adedokun, O.J.; Li, K.; Peyrin-Biroulet, L.; Van Assche, G.; et al. Ustekinumab as Induction and Maintenance Therapy for Ulcerative Colitis. New Engl. J. Med. 2019, 381, 1201–1214. https://doi.org/10.1056/nejmoa1900750.
- Lucaciu, L.A.; Ilieș, M.; Vesa, C.; Seicean, R.; Din, S.; Iuga, C.A.; Seicean, A. Serum Interleukin (IL)-23 and IL-17 Profile in Inflammatory Bowel Disease (IBD) Patients Could Differentiate between Severe and Non-Severe Disease. J. Pers. Med. 2021, 11, 1130. Sewell, G.W.; Kaser, A. Interleukin-23 in the Pathogenesis of Inflammatory Bowel Disease and Implications for Therapeutic Intervention. Crohn’s Colitis 2022, 16, ii3–ii19. https://doi.org/10.1093/ecco-jcc/jjac034.
- Mirsattari, D.; Seyyedmajidi, M.; Zojaji, H.; Haghazali, M.; Orimi, P.G.; Shoushtarizadeh, T.; Almasi, S. The relation between the level of interleukin-23 with duration and severity of ulcerative colitis. Gastroenterol. Hepatol. Bed Bench 2012, 5, 49–53. Hugot, J.-P.; Laurent-Puig, P.; Gower, C.; Olson, J.M.; Lee, J.C.; Beaugerie, L.; Naom, I.; Dupas, J.-L.; Van Gossum, A.; Af, G.D.T.D.; et al. Mapping of a susceptibility locus for Crohn’s disease on chromosome 16. Nature 1996, 379, 821–823. https://doi.org/10.1038/379821a0.
- Oppmann, B.; Lesley, R.; Blom, B.; Timans, J.C.; Xu, Y.; Hunte, B.; Vega, F.; Yu, N.; Wang, J.; Singh, K.; et al. Novel p19 Protein Engages IL-12p40 to Form a Cytokine, IL-23, with Biological Activities Similar as Well as Distinct from IL-12. Immunity 2000, 13, 715–725. Amendola, A.; Butera, A.; Sanchez, M.; Strober, W.; Boirivant, M. Nod2 deficiency is associated with an increased mucosal immunoregulatory response to commensal microorganisms. Mucosal Immunol. 2014, 7, 391–404. https://doi.org/10.1038/mi.2013.58.
- Aggarwal, S.; Ghilardi, N.; Xie, M.-H.; de Sauvage, F.J.; Gurney, A.L. Interleukin-23 Promotes a Distinct CD4 T Cell Activation State Characterized by the Production of Interleukin-17. J. Biol. Chem. 2003, 278, 1910–1914. Barreau, F.; Meinzer, U.; Chareyre, F.; Berrebi, D.; Kawakita, M.; Dussaillant, M.; Foligné, B.; Ollendorff, V.; Heyman, M.; Bonacorsi, S.; et al. CARD15/NOD2 Is Required for Peyer’s Patches Homeostasis in Mice. PLoS ONE 2007, 2, e523. https://doi.org/10.1371/journal.pone.0000523.
- Aden, K.; Rehman, A.; Falk-Paulsen, M.; Secher, T.; Kuiper, J.; Tran, F.; Pfeuffer, S.; Sheibani-Tezerji, R.; Breuer, A.; Luzius, A.; et al. Epithelial IL-23R Signaling Licenses Protective IL-22 Responses in Intestinal Inflammation. Cell Rep. 2016, 16, 2208–2218. Kobayashi, K.S.; Chamaillard, M.; Ogura, Y.; Henegariu, O.; Inohara, N.; Nuñez, G.; Flavell, R.A. Nod2-Dependent Regulation of Innate and Adaptive Immunity in the Intestinal Tract. Science 2005, 307, 731–734. https://doi.org/10.1126/science.1104911.
- Zenewicz, L.A.; Yancopoulos, G.D.; Valenzuela, D.M.; Murphy, A.J.; Stevens, S.; Flavell, R.A. Innate and Adaptive Interleukin-22 Protects Mice from Inflammatory Bowel Disease. Immunity 2008, 29, 947–957. Maeda, S.; Hsu, L.-C.; Liu, H.; Bankston, L.A.; Iimura, M.; Kagnoff, M.F.; Eckmann, L.; Karin, M. Nod2 Mutation in Crohn’s Disease Potentiates NF-κB Activity and IL-1ß Processing. Science 2005, 307, 734–738. https://doi.org/10.1126/science.1103685.
- Sugimoto, K.; Ogawa, A.; Mizoguchi, E.; Shimomura, Y.; Andoh, A.; Bhan, A.K.; Blumberg, R.S.; Xavier, R.J.; Mizoguchi, A. IL-22 ameliorates intestinal inflammation in a mouse model of ulcerative colitis. J. Clin. Investig. 2008, 118, 534–544. Watanabe, T.; Kitani, A.; Murray, P.J.; Wakatsuki, Y.; Fuss, I.J.; Strober, W. Nucleotide Binding Oligomerization Domain 2 Deficiency Leads to Dysregulated TLR2 Signaling and Induction of Antigen-Specific Colitis. Immunity 2006, 25, 473–485. https://doi.org/10.1016/j.immuni.2006.06.018.
- Vlantis, K.; Polykratis, A.; Welz, P.-S.; van Loo, G.; Pasparakis, M.; Wullaert, A. TLR-independent anti-inflammatory function of intestinal epithelial TRAF6 signalling prevents DSS-induced colitis in mice. Gut 2015, 65, 935–943. Yang, Z.; Fuss, I.J.; Watanabe, T.; Asano, N.; Davey, M.P.; Rosenbaum, J.T.; Strober, W.; Kitani, A. NOD2 Transgenic Mice Exhibit Enhanced MDP-Mediated Down-Regulation of TLR2 Responses and Resistance to Colitis Induction. Gastroenterology 2007, 133, 1510–1521. https://doi.org/10.1053/j.gastro.2007.07.025.
- Loftus, E.V., Jr. Clinical epidemiology of inflammatory bowel disease: Incidence, prevalence, and environmental influences. Gastroenterology 2004, 126, 1504–1517. Butera, A.; Di Paola, M.; Pavarini, L.; Strati, F.; Pindo, M.; Sanchez, M.; Cavalieri, D.; Boirivant, M.; De Filippo, C. Nod2 Deficiency in mice is Associated with Microbiota Variation Favouring the Expansion of mucosal CD4+ LAP+ Regulatory Cells. Rep. 2018, 8, 14241. https://doi.org/10.1038/s41598-018-32583-z.
- Heyman, M.B.; Kirschner, B.S.; Gold, B.D.; Ferry, G.; Baldassano, R.; Cohen, S.A.; Winter, H.S.; Fain, P.; King, C.; Smith, T.; et al. Children with early-onset inflammatory bowel disease (IBD): Analysis of a pediatric IBD consortium registry. J. Pediatr. 2005, 146, 35–40. Cohen, L.J.; Cho, J.H.; Gevers, D.; Chu, H. Genetic Factors and the Intestinal Microbiome Guide Development of Microbe-Based Therapies for Inflammatory Bowel Diseases. Gastroenterology 2019, 156, 2174–2189. https://doi.org/10.1053/j.gastro.2019.03.017.
- Zhu, L.; Shi, T.; Zhong, C.; Wang, Y.; Chang, M.; Liu, X. IL-10 and IL-10 Receptor Mutations in Very Early Onset Inflammatory Bowel Disease. Gastroenterol. Res. 2017, 10, 65–69. Hall, A.B.; Tolonen, A.; Xavier, R.J. Human genetic variation and the gut microbiome in disease. Rev. Genet. 2017, 18, 690–699. https://doi.org/10.1038/nrg.2017.63.
- Berg, D.; Davidson, N.; Kühn, R.; Muller, W.; Menon, S.; Holland, G.; Thompson-Snipes, L.; Leach, M.W.; Rennick, D. Enterocolitis and colon cancer in interleukin-10-deficient mice are associated with aberrant cytokine production and CD4(+) TH1-like responses. J. Clin. Investig. 1996, 98, 1010–1020. Hu, S.; Vila, A.V.; Gacesa, R.; Collij, V.; Stevens, C.; Fu, J.M.; Wong, I.; E Talkowski, M.; A Rivas, M.; Imhann, F.; et al. Whole exome sequencing analyses reveal gene–microbiota interactions in the context of IBD. Gut 2021, 70, 285–296. https://doi.org/10.1136/gutjnl-2019-319706.
- Shouval, D.S.; Biswas, A.; Goettel, J.A.; McCann, K.; Conaway, E.; Redhu, N.S.; Mascanfroni, I.D.; Al Adham, Z.; Lavoie, S.; Ibourk, M.; et al. Interleukin-10 Receptor Signaling in Innate Immune Cells Regulates Mucosal Immune Tolerance and Anti-Inflammatory Macrophage Function. Immunity 2014, 40, 706–719. Antoniou, E.; Margonis, G.A.; Angelou, A.; Pikouli, A.; Argiri, P.; Karavokyros, I.; Papalois, A.; Pikoulis, E. The TNBS-induced colitis animal model: An overview. Med. Surg. 2016, 11, 9–15. https://doi.org/10.1016/j.amsu.2016.07.019.
- Spencer, S.D.; Di Marco, F.; Hooley, J.; Pitts-Meek, S.; Bauer, M.; Ryan, A.M.; Sordat, B.; Gibbs, V.C.; Aguet, M. The Orphan Receptor CRF2-4 Is an Essential Subunit of the Interleukin 10 Receptor. J. Exp. Med. 1998, 187, 571–578. Abad, C.; Martinez, C.; Juarranz, M.G.; Arranz, A.; Leceta, J.; Delgado, M.; Gomariz, R.P. Therapeutic effects of vasoactive intestinal peptide in the trinitrobenzene sulfonic acid mice model of Crohn’s disease. Gastroenterology 2003, 124, 961–971. https://doi.org/10.1053/gast.2003.50141.
- Mendoza, J.L.; Schneider, W.M.; Hoffmann, H.-H.; Vercauteren, K.; Jude, K.M.; Xiong, A.; Moraga, I.; Horton, T.M.; Glenn, J.S.; de Jong, Y.P.; et al. The IFN-λ-IFN-λR1-IL-10Rβ Complex Reveals Structural Features Underlying Type III IFN Functional Plasticity. Immunity 2017, 46, 379–392. Laroui, H.; Ingersoll, S.A.; Liu, H.C.; Baker, M.T.; Ayyadurai, S.; Charania, M.A.; Laroui, F.; Yan, Y.; Sitaraman, S.V.; Merlin, D. Dextran Sodium Sulfate (DSS) Induces Colitis in Mice by Forming Nano-Lipocomplexes with Medium-Chain-Length Fatty Acids in the Colon. PLoS ONE 2012, 7, e32084. https://doi.org/10.1371/journal.pone.0032084.
- Sellon, R.K.; Tonkonogy, S.; Schultz, M.; Dieleman, L.A.; Grenther, W.; Balish, E.; Rennick, D.M.; Sartor, R.B. Resident Enteric Bacteria Are Necessary for Development of Spontaneous Colitis and Immune System Activation in Interleukin-10-Deficient Mice. Infect. Immun. 1998, 66, 5224–5231. Kitajima, S.; Takuma, S.; Morimoto, M. Tissue Distribution of Dextran Sulfate Sodium(DSS) in the Acute Phase of Murine DSS-Induced Colitis. Veter- Med Sci. 1999, 61, 67–70. https://doi.org/10.1292/jvms.61.67.
- Colombel, J.-F.; Lémann, M.; Cassagnou, M.; Bouhnik, Y.; Duclos, B.; Dupas, J.-L.; Notteghem, B.; Mary, J.-Y. A Controlled Trial Comparing Ciprofloxacin With Mesalazine for The Treatment of Active Crohn’s Disease. Am. J. Gastroenterol. 1999, 94, 674–678. Chassaing, B.; Aitken, J.D.; Malleshappa, M.; Vijay-Kumar, M. Dextran Sulfate Sodium (DSS)-Induced Colitis in Mice. Protoc. Immunol. 2014, 104, 15.25.1–15.25.14. https://doi.org/10.1002/0471142735.im1525s104.
- Sutherland, L.; Singleton, J.; Sessions, J.; Hanauer, S.; Krawitt, E.; Rankin, G.; Summers, R.; Mekhjian, H.; Greenberger, N.; Kelly, M. Double blind, placebo controlled trial of metronidazole in Crohn’s disease. Gut 1991, 32, 1071–1075. Perše, M.; Cerar, A. Dextran Sodium Sulphate Colitis Mouse Model: Traps and Tricks. Biomed. Biotechnol. 2012, 2012, 718617. https://doi.org/10.1155/2012/718617.
- Madsen, K.L.; Doyle, J.S.; Tavernini, M.M.; Jewell, L.D.; Rennie, R.P.; Fedorak, R.N. Antibiotic therapy attenuates colitis in interleukin 10 gene–deficient mice. Gastroenterology 2000, 118, 1094–1105. Kim, S.H.; Kwon, D.; Son, S.W.; Bin Jeong, T.; Lee, S.; Kwak, J.-H.; Cho, J.-Y.; Hwang, D.Y.; Seo, M.-S.; Kim, K.S.; et al. Inflammatory responses of C57BL/6NKorl mice to dextran sulfate sodium-induced colitis: Comparison between three C57BL/6 N sub-strains. Anim. Res. 2021, 37, 8. https://doi.org/10.1186/s42826-021-00084-2.
- Gunasekera, D.C.; Ma, J.; Vacharathit, V.; Shah, P.; Ramakrishnan, A.; Uprety, P.; Shen, Z.; Sheh, A.; Brayton, C.F.; Whary, M.T.; et al. The development of colitis in Il10−/− mice is dependent on IL-22. Mucosal Immunol. 2020, 13, 493–506. A Dieleman, L.; Palmen, M.J.H.J.; Akol, H.; Bloemena, E.; Peña, A.S.; Meuwissen, S.G.M.; Van Rees, E.P. Chronic experimental colitis induced by dextran sulphate sodium (DSS) is characterized by Th1 and Th2 cytokines. Exp. Immunol. 1998, 114, 385–391. https://doi.org/10.1046/j.1365-2249.1998.00728.x.
- Ott, S.J.; Musfeldt, M.; Wenderoth, D.F.; Hampe, J.; Brant, O.; Fölsch, U.R.; Timmis, K.N.; Schreiber, S. Reduction in diversity of the colonic mucosa associated bacterial microflora in patients with active inflammatory bowel disease. Gut 2004, 53, 685–693. Stevceva, L.; Pavli, P.; Husband, A.; Ramsay, A.; Doe, W. Dextran sulphate sodium-induced colitis is ameliorated in interleukin 4 deficient mice. Genes Immun. 2001, 2, 309–316. https://doi.org/10.1038/sj.gene.6363782.
- Aranda, R.; Sydora, B.C.; McAllister, P.L.; Binder, S.W.; Yang, H.Y.; Targan, S.R.; Kronenberg, M. Analysis of intestinal lymphocytes in mouse colitis mediated by transfer of CD4+, CD45RBhigh T cells to SCID recipients. J. Immunol. 1997, 158, 3464–3473. Melgar, S.; Karlsson, A.; Michaëlsson, E. Acute colitis induced by dextran sulfate sodium progresses to chronicity in C57BL/6 but not in BALB/c mice: Correlation between symptoms and inflammation. J. Physiol. Liver Physiol. 2005, 288, G1328–G1338. https://doi.org/10.1152/ajpgi.00467.2004.
- Feng, T.; Wang, L.; Schoeb, T.R.; Elson, C.O.; Cong, Y. Microbiota innate stimulation is a prerequisite for T cell spontaneous proliferation and induction of experimental colitis. J. Exp. Med. 2010, 207, 1321–1332. Dieleman, L.A.; Ridwan, B.U.; Tennyson, G.S.; Beagley, K.W.; Bucy, R.; Elson, C.O. Dextran sulfate sodium-induced colitis occurs in severe combined immunodeficient mice. Gastroenterology 1994, 107, 1643–1652. https://doi.org/10.1016/0016-5085(94)90803-6.
- Mottet, C.; Uhlig, H.H.; Powrie, F. Cutting Edge: Cure of Colitis by CD4+CD25+ Regulatory T Cells. J. Immunol. 2003, 170, 3939–3943. Kim, T.W.; Seo, J.N.; Suh, Y.H.; Park, H.J.; Kim, J.H.; Kim, J.Y.; Oh, K.I. Involvement of lymphocytes in dextran sulfate sodium-induced experimental colitis. World J. Gastroenterol. 2006, 12, 302–305. https://doi.org/10.3748/wjg.v12.i2.302.
- Kjellev, S.; Lundsgaard, D.; Poulsen, S.S.; Markholst, H. Reconstitution of Scid mice with CD4+CD25− T cells leads to rapid colitis: An improved model for pharmacologic testing. Int. Immunopharmacol. 2006, 6, 1341–1354. Tanaka, M.; Riddell, R.H.; Saito, H.; Soma, Y.; Hidaka, H.; Kudo, H. Morphologic Criteria Applicable to Biopsy Specimens for Effective Distinction of Inflammatory Bowel Disease from Other Forms of Colitis and of Crohn’s Disease from Ulcerative Colitis. J. Gastroenterol. 1999, 34, 55–67. https://doi.org/10.1080/00365529950172844.
- Powrie, F.; Leach, M.W.; Mauze, S.; Menon, S.; Caddle, L.B.; Coffman, R.L. Inhibition of Thl responses prevents inflammatory bowel disease in scid mice reconstituted with CD45RBhi CD4+ T cells. Immunity 1994, 1, 553–562. Fujino, S.; Andoh, A.; Bamba, S.; Ogawa, A.; Hata, K.; Araki, Y.; Bamba, T.; Fujiyama, Y. Increased expression of interleukin 17 in inflammatory bowel disease. Gut 2003, 52, 65–70. https://doi.org/10.1136/gut.52.1.65.
- Ahern, P.P.; Schiering, C.; Buonocore, S.; McGeachy, M.J.; Cua, D.J.; Maloy, K.J.; Powrie, F. Interleukin-23 Drives Intestinal Inflammation through Direct Activity on T Cells. Immunity 2010, 33, 279–288. Gheita, T.A.; El Gazzar, I.I.; El-Fishawy, H.S.; Aboul-Ezz, M.A.; Kenawy, S.A. Involvement of IL-23 in enteropathic arthritis patients with inflammatory bowel disease: Preliminary results. Rheumatol. 2014, 33, 713–717. https://doi.org/10.1007/s10067-013-2469-y.
- Hue, S.; Ahern, P.; Buonocore, S.; Kullberg, M.C.; Cua, D.J.; McKenzie, B.S.; Powrie, F.; Maloy, K.J. Interleukin-23 drives innate and T cell–mediated intestinal inflammation. J. Exp. Med. 2006, 203, 2473–2483. Jiang, W.; Su, J.; Zhang, X.; Cheng, X.; Zhou, J.; Shi, R.; Zhang, H. Elevated levels of Th17 cells and Th17-related cytokines are associated with disease activity in patients with inflammatory bowel disease. Agents Actions 2014, 63, 943–950. https://doi.org/10.1007/s00011-014-0768-7.
- Brasseit, J.; Chung, C.K.C.K.; Noti, M.; Zysset, D.; Hoheisel-Dickgreber, N.; Genitsch, V.; Corazza, N.; Mueller, C. Divergent Roles of Interferon-γ and Innate Lymphoid Cells in Innate and Adaptive Immune Cell-Mediated Intestinal Inflammation. Front. Immunol. 2018, 9, 23. Lucaciu, L.A.; Ilieș, M.; Vesa, C.; Seicean, R.; Din, S.; Iuga, C.A.; Seicean, A. Serum Interleukin (IL)-23 and IL-17 Profile in Inflammatory Bowel Disease (IBD) Patients Could Differentiate between Severe and Non-Severe Disease. Pers. Med. 2021, 11, 1130. https://doi.org/10.3390/jpm11111130.
- Harbour, S.N.; Maynard, C.L.; Zindl, C.L.; Schoeb, T.R.; Weaver, C.T. Th17 cells give rise to Th1 cells that are required for the pathogenesis of colitis. Proc. Natl. Acad. Sci. USA 2015, 112, 7061–7066. Mirsattari, D.; Seyyedmajidi, M.; Zojaji, H.; Haghazali, M.; Orimi, P.G.; Shoushtarizadeh, T.; Almasi, S. The relation between the level of interleukin-23 with duration and severity of ulcerative colitis. Hepatol. bed bench 2012, 5, 49–53.
- Poholek, C.; Dulson, S.J.; Zajac, A.J.; Harrington, L.E. IL-21 Controls ILC3 Cytokine Production and Promotes a Protective Phenotype in a Mouse Model of Colitis. ImmunoHorizons 2019, 3, 194–202. Oppmann, B.; Lesley, R.; Blom, B.; Timans, J.C.; Xu, Y.; Hunte, B.; Vega, F.; Yu, N.; Wang, J.; Singh, K.; et al. Novel p19 Protein Engages IL-12p40 to Form a Cytokine, IL-23, with Biological Activities Similar as Well as Distinct from IL-12. Immunity 2000, 13, 715–725. https://doi.org/10.1016/s1074-7613(00)00070-4.
- Bosma, G.C.; Fried, M.; Custer, R.P.; Carroll, A.; Gibson, D.M.; Bosma, M.J. Evidence of functional lymphocytes in some (leaky) scid mice. J. Exp. Med. 1988, 167, 1016–1033. Aggarwal, S.; Ghilardi, N.; Xie, M.-H.; de Sauvage, F.J.; Gurney, A.L. Interleukin-23 Promotes a Distinct CD4 T Cell Activation State Characterized by the Production of Interleukin-17. Biol. Chem. 2003, 278, 1910–1914. https://doi.org/10.1074/jbc.m207577200.
- Karo, J.M.; Schatz, D.G.; Sun, J.C. The RAG Recombinase Dictates Functional Heterogeneity and Cellular Fitness in Natural Killer Cells. Cell 2014, 159, 94–107. Aden, K.; Rehman, A.; Falk-Paulsen, M.; Secher, T.; Kuiper, J.; Tran, F.; Pfeuffer, S.; Sheibani-Tezerji, R.; Breuer, A.; Luzius, A.; et al. Epithelial IL-23R Signaling Licenses Protective IL-22 Responses in Intestinal Inflammation. Cell Rep. 2016, 16, 2208–2218. https://doi.org/10.1016/j.celrep.2016.07.054.
- Kwan, A.; Abraham, R.S.; Currier, R.; Brower, A.; Andruszewski, K.; Abbott, J.; Baker, M.; Ballow, M.; Bartoshesky, L.E.; Bonilla, F.A.; et al. Newborn Screening for Severe Combined Immunodeficiency in 11 Screening Programs in the United States. JAMA 2014, 312, 729–738. Zenewicz, L.A.; Yancopoulos, G.D.; Valenzuela, D.M.; Murphy, A.J.; Stevens, S.; Flavell, R.A. Innate and Adaptive Interleukin-22 Protects Mice from Inflammatory Bowel Disease. Immunity 2008, 29, 947–957. https://doi.org/10.1016/j.immuni.2008.11.003.
- Sugimoto, K.; Ogawa, A.; Mizoguchi, E.; Shimomura, Y.; Andoh, A.; Bhan, A.K.; Blumberg, R.S.; Xavier, R.J.; Mizoguchi, A. IL-22 ameliorates intestinal inflammation in a mouse model of ulcerative colitis. Clin. Investig. 2008, 118, 534–544. https://doi.org/10.1172/jci33194.
- Kobayashi, T.; Okamoto, S.; Hisamatsu, T.; Kamada, N.; Chinen, H.; Saito, R.; Kitazume, M.T.; Nakazawa, A.; Sugita, A.; Koganei, K.; et al. IL23 differentially regulates the Th1/Th17 balance in ulcerative colitis and Crohn’s disease. Gut 2008, 57, 1682–1689. https://doi.org/10.1136/gut.2007.135053.
- Gaffen, S.L. Structure and signalling in the IL-17 receptor family. Rev. Immunol. 2009, 9, 556–567. https://doi.org/10.1038/nri2586.
- Geremia, A.; Arancibia-Cárcamo, C.V. Innate Lymphoid Cells in Intestinal Inflammation. Immunol. 2017, 8, 1296. https://doi.org/10.3389/fimmu.2017.01296.
- Caruso, R.; Fina, D.; Peluso, I.; Stolfi, C.; Fantini, M.C.; Gioia, V.; Caprioli, F.; Blanco, G.D.V.; Paoluzi, O.A.; MacDonald, T.T.; et al. A Functional Role for Interleukin-21 in Promoting the Synthesis of the T-Cell Chemoattractant, MIP-3α, by Gut Epithelial Cells. Gastroenterology 2007, 132, 166–175. https://doi.org/10.1053/j.gastro.2006.09.053.
- Kim, Y.S.; Lee, M.H.; Ju, A.S.; Rhee, K.-J. Th17 Responses Are Not Induced in Dextran Sodium Sulfate Model of Acute Colitis. Immune Netw. 2011, 11, 416–419. https://doi.org/10.4110/in.2011.11.6.416.
- Ogawa, A.; Andoh, A.; Araki, Y.; Bamba, T.; Fujiyama, Y. Neutralization of interleukin-17 aggravates dextran sulfate sodium-induced colitis in mice. Immunol. 2004, 110, 55–62. https://doi.org/10.1016/j.clim.2003.09.013.
- Lee, J.S.; Tato, C.M.; Joyce-Shaikh, B.; Gulen, M.F.; Cayatte, C.; Chen, Y.; Blumenschein, W.M.; Judo, M.; Ayanoglu, G.; McClanahan, T.K.; et al. Interleukin-23-Independent IL-17 Production Regulates Intestinal Epithelial Permeability. Immunity 2015, 43, 727–738. https://doi.org/10.1016/j.immuni.2015.09.003.
- Ito, R.; Kita, M.; Shin-Ya, M.; Kishida, T.; Urano, A.; Takada, R.; Sakagami, J.; Imanishi, J.; Iwakura, Y.; Okanoue, T.; et al. Involvement of IL-17A in the pathogenesis of DSS-induced colitis in mice. Biophys. Res. Commun. 2008, 377, 12–16. https://doi.org/10.1016/j.bbrc.2008.09.019.
- Tang, C.; Kakuta, S.; Shimizu, K.; Kadoki, M.; Kamiya, T.; Shimazu, T.; Kubo, S.; Saijo, S.; Ishigame, H.; Nakae, S.; et al. Suppression of IL-17F, but not of IL-17A, provides protection against colitis by inducing Treg cells through modification of the intestinal microbiota. Immunol. 2018, 19, 755–765. https://doi.org/10.1038/s41590-018-0134-y.
- Chen, Y.; Chen, Y.; Cao, P.; Su, W.; Zhan, N.; Dong, W. Fusobacterium nucleatum facilitates ulcerative colitis through activating IL-17F signaling to NF-κB via the upregulation of CARD3 expression. Pathol. 2020, 250, 170–182. https://doi.org/10.1002/path.5358.
- Hueber, W.; E Sands, B.; Lewitzky, S.; Vandemeulebroecke, M.; Reinisch, W.; Higgins, P.D.R.; Wehkamp, J.; Feagan, B.G.; Yao, M.D.; Karczewski, M.; et al. Secukinumab, a human anti-IL-17A monoclonal antibody, for moderate to severe Crohn’s disease: Unexpected results of a randomised, double-blind placebo-controlled trial. Gut 2012, 61, 1693–1700. https://doi.org/10.1136/gutjnl-2011-301668.
- Targan, S.R.; Feagan, B.; Vermeire, S.; Panaccione, R.; Melmed, G.Y.; Landers, C.; Li, D.; Russell, C.; Newmark, R.; Zhang, N.; et al. A Randomized, Double-Blind, Placebo-Controlled Phase 2 Study of Brodalumab in Patients With Moderate-to-Severe Crohn’s Disease. J. Gastroenterol. 2016, 111, 1599–1607. https://doi.org/10.1038/ajg.2016.298.
- Glatt, S.; Helmer, E.; Haier, B.; Strimenopoulou, F.; Price, G.; Vajjah, P.; Harari, O.A.; Lambert, J.; Shaw, S. First-in-human randomized study of bimekizumab, a humanized monoclonal antibody and selective dual inhibitor of IL-17A and IL-17F, in mild psoriasis. J. Clin. Pharmacol. 2017, 83, 991–1001. https://doi.org/10.1111/bcp.13185.
- Gordon, K.B.; Foley, P.; Krueger, J.G.; Pinter, A.; Reich, K.; Vender, R.; Vanvoorden, V.; Madden, C.; White, K.; Cioffi, C.; et al. Bimekizumab efficacy and safety in moderate to severe plaque psoriasis (BE READY): A multicentre, double-blind, placebo-controlled, randomised withdrawal phase 3 trial. Lancet 2021, 397, 475–486. https://doi.org/10.1016/s0140-6736(21)00126-4.
- Reimund, J.M.; Wittersheim, C.; Dumont, S.; Muller, C.D.; Kenney, J.S.; Baumann, R.; Poindron, P.; Duclos, B. Increased production of tumour necrosis factor-alpha interleukin-1 beta, and interleukin-6 by morphologically normal intestinal biopsies from patients with Crohn’s disease. Gut 1996, 39, 684–689. https://doi.org/10.1136/gut.39.5.684.
- Kontoyiannis, D.; Pasparakis, M.; Pizarro, T.T.; Cominelli, F.; Kollias, G. Impaired On/Off Regulation of TNF Biosynthesis in Mice Lacking TNF AU-Rich Elements: Implications for Joint and Gut-Associated Immunopathologies. Immunity 1999, 10, 387–398. https://doi.org/10.1016/s1074-7613(00)80038-2.
- Kojouharoff, G.; Hans, W.; Obermeier, F.; Ma¨nnel, D.N.; Andus, T.; Scho¨lmerich, J.; Gross, V.; Falk, W. Neutralization of tumour necrosis factor (TNF) but not of IL-1 reduces inflammation in chronic dextran sulphate sodium-induced colitis in mice. Exp. Immunol. 1997, 107, 353–358. https://doi.org/10.1111/j.1365-2249.1997.291-ce1184.x.
- Wang, K.; Diao, L.-H.; Gong, Y.; Liu, X.; Li, Y. NEMO differentially regulates TCR and TNF-α induced NF-κB pathways and has an inhibitory role in TCR-induced NF-κB activation. Signal. 2012, 24, 1556–1564. https://doi.org/10.1016/j.cellsig.2012.03.022.
- Targan, S.R.; Hanauer, S.B.; Van Deventer, S.J.; Mayer, L.; Present, D.H.; Braakman, T.; DeWoody, K.L.; Schaible, T.F.; Rutgeerts, P.J. A Short-Term Study of Chimeric Monoclonal Antibody cA2 to Tumor Necrosis Factor α for Crohn’s Disease. Engl. J. Med. 1997, 337, 1029–1036. https://doi.org/10.1056/nejm199710093371502.
- Kopylov, U.; Seidman, E. Predicting durable response or resistance to antitumor necrosis factor therapy in inflammatory bowel disease. Adv. Gastroenterol. 2016, 9, 513–526. https://doi.org/10.1177/1756283x16638833.
- Dahmus, J.; Rosario, M.; Clarke, K. Risk of Lymphoma Associated with Anti-TNF Therapy in Patients with Inflammatory Bowel Disease: Implications for Therapy. Exp. Gastroenterol. 2020, 13, 339–350. https://doi.org/10.2147/ceg.s237646.
- Schierova, D.; Roubalova, R.; Kolar, M.; Stehlikova, Z.; Rob, F.; Jackova, Z.; Coufal, S.; Thon, T.; Mihula, M.; Modrak, M.; et al. Fecal Microbiome Changes and Specific Anti-Bacterial Response in Patients with IBD during Anti-TNF Therapy. Cells 2021, 10, 3188. https://doi.org/10.3390/cells10113188.
- Vlantis, K.; Polykratis, A.; Welz, P.-S.; van Loo, G.; Pasparakis, M.; Wullaert, A. TLR-independent anti-inflammatory function of intestinal epithelial TRAF6 signalling prevents DSS-induced colitis in mice. Gut 2015, 65, 935–943. https://doi.org/10.1136/gutjnl-2014-308323.
- Fellermann, K.; Stange, D.E.; Schaeffeler, E.; Schmalzl, H.; Wehkamp, J.; Bevins, C.L.; Reinisch, W.; Teml, A.; Schwab, M.; Lichter, P.; et al. A Chromosome 8 Gene-Cluster Polymorphism with Low Human Beta-Defensin 2 Gene Copy Number Predisposes to Crohn Disease of the Colon. J. Hum. Genet. 2006, 79, 439–448. https://doi.org/10.1086/505915.
- Vlantis, K.; Wullaert, A.; Polykratis, A.; Kondylis, V.; Dannappel, M.; Schwarzer, R.; Welz, P.; Corona, T.; Walczak, H.; Weih, F.; et al. NEMO Prevents RIP Kinase 1-Mediated Epithelial Cell Death and Chronic Intestinal Inflammation by NF-κB-Dependent and -Independent Functions. Immunity 2016, 44, 553–567. https://doi.org/10.1016/j.immuni.2016.02.020.
- Kohn, L.L.; Braun, M.; Cordoro, K.M.; McCalmont, T.H.; Shah, S.D.; Frieden, I.J.; Mathur, A.N. Skin and Mucosal Manifestations in NEMO Syndrome: A Case Series and Literature Review. Dermatol. 2022, 39, 84–90. https://doi.org/10.1111/pde.14905.
- Loftus, E.V., Jr. Clinical epidemiology of inflammatory bowel disease: Incidence, prevalence, and environmental influences. Gastroenterology 2004, 126, 1504–1517. https://doi.org/10.1053/j.gastro.2004.01.063.
- Heyman, M.B.; Kirschner, B.S.; Gold, B.D.; Ferry, G.; Baldassano, R.; Cohen, S.A.; Winter, H.S.; Fain, P.; King, C.; Smith, T.; et al. Children with early-onset inflammatory bowel disease (IBD): Analysis of a pediatric IBD consortium registry. Pediatr. 2005, 146, 35–40. https://doi.org/10.1016/j.jpeds.2004.08.043.
- Zhu, L.; Shi, T.; Zhong, C.; Wang, Y.; Chang, M.; Liu, X. IL-10 and IL-10 Receptor Mutations in Very Early Onset Inflammatory Bowel Disease. Res. 2017, 10, 65–69. https://doi.org/10.14740/gr740w.
- Berg, D.; Davidson, N.; Kühn, R.; Muller, W.; Menon, S.; Holland, G.; Thompson-Snipes, L.; Leach, M.W.; Rennick, D. Enterocolitis and colon cancer in interleukin-10-deficient mice are associated with aberrant cytokine production and CD4(+) TH1-like responses. Clin. Investig. 1996, 98, 1010–1020. https://doi.org/10.1172/jci118861.
- Shouval, D.S.; Biswas, A.; Goettel, J.A.; McCann, K.; Conaway, E.; Redhu, N.S.; Mascanfroni, I.D.; Al Adham, Z.; Lavoie, S.; Ibourk, M.; et al. Interleukin-10 Receptor Signaling in Innate Immune Cells Regulates Mucosal Immune Tolerance and Anti-Inflammatory Macrophage Function. Immunity 2014, 40, 706–719. https://doi.org/10.1016/j.immuni.2014.03.011.
- Spencer, S.D.; Di Marco, F.; Hooley, J.; Pitts-Meek, S.; Bauer, M.; Ryan, A.M.; Sordat, B.; Gibbs, V.C.; Aguet, M. The Orphan Receptor CRF2-4 Is an Essential Subunit of the Interleukin 10 Receptor. Exp. Med. 1998, 187, 571–578. https://doi.org/10.1084/jem.187.4.571.
- Mendoza, J.L.; Schneider, W.M.; Hoffmann, H.-H.; Vercauteren, K.; Jude, K.M.; Xiong, A.; Moraga, I.; Horton, T.M.; Glenn, J.S.; de Jong, Y.P.; et al. The IFN-λ-IFN-λR1-IL-10Rβ Complex Reveals Structural Features Underlying Type III IFN Functional Plasticity. Immunity 2017, 46, 379–392. https://doi.org/10.1016/j.immuni.2017.02.017.
- Yen, D.; Cheung, J.; Scheerens, H.; Poulet, F.; McClanahan, T.; McKenzie, B.; Kleinschek, M.A.; Owyang, A.; Mattson, J.; Blumenschein, W.; et al. IL-23 is essential for T cell–mediated colitis and promotes inflammation via IL-17 and IL-6. Clin. Investig. 2006, 116, 1310–1316. https://doi.org/10.1172/jci21404.
- Eyerich, K.; DiMartino, V.; Cavani, A. IL-17 and IL-22 in immunity: Driving protection and pathology. J. Immunol. 2017, 47, 607–614. https://doi.org/10.1002/eji.201646723.
- Gu, Y.; Yang, J.; Ouyang, X.; Liu, W.; Li, H.; Yang, J.; Bromberg, J.; Chen, S.; Mayer, L.; Unkeless, J.C.; et al. Interleukin 10 suppresses Th17 cytokines secreted by macrophages and T cells. J. Immunol. 2008, 38, 1807–1813. https://doi.org/10.1002/eji.200838331.
- Gunasekera, D.C.; Ma, J.; Vacharathit, V.; Shah, P.; Ramakrishnan, A.; Uprety, P.; Shen, Z.; Sheh, A.; Brayton, C.F.; Whary, M.T.; et al. The development of colitis in Il10−/− mice is dependent on IL-22. Mucosal Immunol. 2020, 13, 493–506. https://doi.org/10.1038/s41385-019-0252-3.
- Sellon, R.K.; Tonkonogy, S.; Schultz, M.; Dieleman, L.A.; Grenther, W.; Balish, E.; Rennick, D.M.; Sartor, R.B. Resident Enteric Bacteria Are Necessary for Development of Spontaneous Colitis and Immune System Activation in Interleukin-10-Deficient Mice. Immun. 1998, 66, 5224–5231. https://doi.org/10.1128/iai.66.11.5224-5231.1998.
- Colombel, J.-F.; Lémann, M.; Cassagnou, M.; Bouhnik, Y.; Duclos, B.; Dupas, J.-L.; Notteghem, B.; Mary, J.-Y. A Controlled Trial Comparing Ciprofloxacin With Mesalazine for The Treatment of Active Crohn’s Disease. J. Gastroenterol. 1999, 94, 674–678. https://doi.org/10.1111/j.1572-0241.1999.935_q.x.
- Sutherland, L.; Singleton, J.; Sessions, J.; Hanauer, S.; Krawitt, E.; Rankin, G.; Summers, R.; Mekhjian, H.; Greenberger, N.; Kelly, M. Double blind, placebo controlled trial of metronidazole in Crohn’s disease. Gut 1991, 32, 1071–1075. https://doi.org/10.1136/gut.32.9.1071.
- Madsen, K.L.; Doyle, J.S.; Tavernini, M.M.; Jewell, L.D.; Rennie, R.P.; Fedorak, R.N. Antibiotic therapy attenuates colitis in interleukin 10 gene–deficient mice. Gastroenterology 2000, 118, 1094–1105. https://doi.org/10.1016/s0016-5085(00)70362-3.
- Ott, S.J.; Musfeldt, M.; Wenderoth, D.F.; Hampe, J.; Brant, O.; Fölsch, U.R.; Timmis, K.N.; Schreiber, S. Reduction in diversity of the colonic mucosa associated bacterial microflora in patients with active inflammatory bowel disease. Gut 2004, 53, 685–693. https://doi.org/10.1136/gut.2003.025403.
- Burich, A.; Hershberg, R.; Waggie, K.; Zeng, W.; Brabb, T.; Westrich, G.; Viney, J.L.; Maggio-Price, L. Helicobacter-induced inflammatory bowel disease in IL-10- and T cell-deficient mice. J. Physiol. Liver Physiol. 2001, 281, G764–G778. https://doi.org/10.1152/ajpgi.2001.281.3.g764.
- Kullberg, M.C.; Ward, J.M.; Gorelick, P.L.; Caspar, P.; Hieny, S.; Cheever, A.; Jankovic, D.; Sher, A. Helicobacter hepaticus Triggers Colitis in Specific-Pathogen-Free Interleukin-10 (IL-10)-Deficient Mice through an IL-12- and Gamma Interferon-Dependent Mechanism. Immun. 1998, 66, 5157–5166. https://doi.org/10.1128/iai.66.11.5157-5166.1998.
- Balish, E.; Warner, T. Enterococcus faecalis Induces Inflammatory Bowel Disease in Interleukin-10 Knockout Mice. J. Pathol. 2002, 160, 2253–2257. https://doi.org/10.1016/s0002-9440(10)61172-8.
- Kim, S.C.; Tonkonogy, S.L.; Albright, C.A.; Tsang, J.; Balish, E.J.; Braun, J.; Huycke, M.M.; Sartor, R.B. Variable phenotypes of enterocolitis in interleukin 10-deficient mice monoassociated with two different commensal bacteria. Gastroenterology 2005, 128, 891–906. https://doi.org/10.1053/j.gastro.2005.02.009.
- Madsen, K.L.; Doyle, J.S.; Jewell, L.D.; Tavernini, M.M.; Fedorak, R.N. Lactobacillus species prevents colitis in interleukin 10 gene–deficient mice. Gastroenterology 1999, 116, 1107–1114. https://doi.org/10.1016/s0016-5085(99)70013-2.
- Madsen, K.; Cornish, A.; Soper, P.; McKaigney, C.; Jijon, H.; Yachimec, C.; Doyle, J.; Jewell, L.; De Simone, C. Probiotic bacteria enhance murine and human intestinal epithelial barrier function. Gastroenterology 2001, 121, 580–591. https://doi.org/10.1053/gast.2001.27224.
- Li, S.; Jin, Y.; Fu, W.; Cox, A.D.; Lee, D.; Reddivari, L. Intermittent antibiotic treatment accelerated the development of colitis in IL-10 knockout mice. Pharmacother. 2022, 146, 112486. https://doi.org/10.1016/j.biopha.2021.112486.
- van Deventer, S.; Elson, C.; Fedorak, R. Multiple doses of intravenous interleukin 10 in steroid-refractory Crohn’s disease. Crohn’s Disease Study Group. Gastroenterology 1997, 113, 383–389. https://doi.org/10.1053/gast.1997.v113.pm9247454.
- Fedorak, R.N.; Gangl, A.; Elson, C.O.; Rutgeerts, P.; Schreiber, S.; Wild, G.; Hanauer, S.B.; Kilian, A.; Cohard, M.; LeBeaut, A.; et al. Recombinant human interleukin 10 in the treatment of patients with mild to moderately active Crohn’s disease. . Gastroenterology 2000, 119, 1473–1482. https://doi.org/10.1053/gast.2000.20229.
- Huhn, R.D.; Radwanski, E.; Gallo, J.; Affrime, M.B.; Sabo, R.; Gonyo, G.; Monge, A.; Cutler, D.L. Pharmacodynamics of subcutaneous recombinant human interleukin-10 in healthy volunteers*. Pharmacol. Ther. 1997, 62, 171–180. https://doi.org/10.1016/s0009-9236(97)90065-5.
- Schreiber, S.; Fedorak, R.N.; Nielsen, O.H.; Wild, G.; Williams, C.; Nikolaus, S.; Jacyna, M.; Lashner, B.A.; Gangl, A.; Rutgeerts, P.; et al. Safety and efficacy of recombinant human interleukin 10 in chronic active Crohn’s disease. Gastroenterology 2000, 119, 1461–1472. https://doi.org/10.1053/gast.2000.20196.
- Sosman, J.A.; Verma, A.; Moss, S.; Sorokin, P.; Blend, M.; Bradlow, B.; Chachlani, N.; Cutler, D.; Sabo, R.; Nelson, M.; et al. Interleukin 10-induced thrombocytopenia in normal healthy adult volunteers: Evidence for decreased platelet production. J. Haematol. 2000, 111, 104–111. https://doi.org/10.1046/j.1365-2141.2000.02314.x.
- Tilg, H.; Van Montfrans, C.; Ende, A.V.D.; Kaser, A.; Van Deventer, S.J.H.; Schreiber, S.; Gregor, M.; Ludwiczek, O.; Rutgeerts, P.; Gasche, C.; et al. Treatment of Crohn’s disease with recombinant human interleukin 10 induces the proinflammatory cytokine interferon gamma. Gut 2002, 50, 191–195. https://doi.org/10.1136/gut.50.2.191.
- Steidler, L.; Hans, W.; Schotte, L.; Neirynck, S.; Obermeier, F.; Falk, W.; Fiers, W.; Remaut, E. Treatment of Murine Colitis by Lactococcus lactis Secreting Interleukin-10. Science 2000, 289, 1352–1355. https://doi.org/10.1126/science.289.5483.1352.
- Zurita-Turk, M.; Souza, B.M.; De Castro, C.P.; Pereira, V.B.; Da Cunha, V.P.; Preisser, T.M.; De Faria, A.M.C.; Machado, D.C.C.; Miyoshi, A. Attenuation of intestinal inflammation in IL-10 deficient mice by a plasmid carrying Lactococcus lactis strain. BMC Biotechnol. 2020, 20, 38. https://doi.org/10.1186/s12896-020-00631-0.
- Fay, N.C.; Muthusamy, B.-P.; Nyugen, L.P.; Desai, R.C.; Taverner, A.; Mackay, J.; Seung, M.; Hunter, T.; Liu, K.; Chandalia, A.; et al. A Novel Fusion of IL-10 Engineered to Traffic across Intestinal Epithelium to Treat Colitis. Immunol. 2020, 205, 3191–3204. https://doi.org/10.4049/jimmunol.2000848.
- Braat, H.; Rottiers, P.; Hommes, D.W.; Huyghebaert, N.; Remaut, E.; Remon, J.; van Deventer, S.J.; Neirynck, S.; Peppelenbosch, M.P.; Steidler, L. A Phase I Trial With Transgenic Bacteria Expressing Interleukin-10 in Crohn’s Disease. Gastroenterol. Hepatol. 2006, 4, 754–759. https://doi.org/10.1016/j.cgh.2006.03.028.
- Kucharzik, T.; Stoll, R.; Lügering, N.; Domschke, W. Circulating antiinflammatory cytokine IL-10 in patients with inflammatory bowel disease (IBD). Exp. Immunol. 1995, 100, 452–456. https://doi.org/10.1111/j.1365-2249.1995.tb03721.x.
- Colombel, J.-F.; Rutgeerts, P.; Malchow, H.; Jacyna, M.; Nielsen, O.H.; Rask-Madsen, J.; Van Deventer, S.; Ferguson, A.; Desreumaux, P.; Forbes, A.; et al. Interleukin 10 (Tenovil) in the prevention of postoperative recurrence of Crohn’s disease. Gut 2001, 49, 42–46. https://doi.org/10.1136/gut.49.1.42.
- Aranda, R.; Sydora, B.C.; McAllister, P.L.; Binder, S.W.; Yang, H.Y.; Targan, S.R.; Kronenberg, M. Analysis of intestinal lymphocytes in mouse colitis mediated by transfer of CD4+, CD45RBhigh T cells to SCID recipients. Immunol. 1997, 158, 3464–3473.
- Feng, T.; Wang, L.; Schoeb, T.R.; Elson, C.O.; Cong, Y. Microbiota innate stimulation is a prerequisite for T cell spontaneous proliferation and induction of experimental colitis. Exp. Med. 2010, 207, 1321–1332. https://doi.org/10.1084/jem.20092253.
- Mottet, C.; Uhlig, H.H.; Powrie, F. Cutting Edge: Cure of Colitis by CD4+CD25+ Regulatory T Cells. Immunol. 2003, 170, 3939–3943. https://doi.org/10.4049/jimmunol.170.8.3939.
- Kjellev, S.; Lundsgaard, D.; Poulsen, S.S.; Markholst, H. Reconstitution of Scid mice with CD4+CD25− T cells leads to rapid colitis: An improved model for pharmacologic testing. Immunopharmacol. 2006, 6, 1341–1354. https://doi.org/10.1016/j.intimp.2006.04.017.
- Powrie, F.; Leach, M.W.; Mauze, S.; Menon, S.; Caddle, L.B.; Coffman, R.L. Inhibition of Thl responses prevents inflammatory bowel disease in scid mice reconstituted with CD45RBhi CD4+ T cells. Immunity 1994, 1, 553–562. https://doi.org/10.1016/1074-7613(94)90045-0.
- Ahern, P.P.; Schiering, C.; Buonocore, S.; McGeachy, M.J.; Cua, D.J.; Maloy, K.J.; Powrie, F. Interleukin-23 Drives Intestinal Inflammation through Direct Activity on T Cells. Immunity 2010, 33, 279–288. https://doi.org/10.1016/j.immuni.2010.08.010.
- Hue, S.; Ahern, P.; Buonocore, S.; Kullberg, M.C.; Cua, D.J.; McKenzie, B.S.; Powrie, F.; Maloy, K.J. Interleukin-23 drives innate and T cell–mediated intestinal inflammation. Exp. Med. 2006, 203, 2473–2483. https://doi.org/10.1084/jem.20061099.
- Brasseit, J.; Chung, C.K.C.K.; Noti, M.; Zysset, D.; Hoheisel-Dickgreber, N.; Genitsch, V.; Corazza, N.; Mueller, C. Divergent Roles of Interferon-γ and Innate Lymphoid Cells in Innate and Adaptive Immune Cell-Mediated Intestinal Inflammation. Immunol. 2018, 9, 23. https://doi.org/10.3389/fimmu.2018.00023.
- Harbour, S.N.; Maynard, C.L.; Zindl, C.L.; Schoeb, T.R.; Weaver, C.T. Th17 cells give rise to Th1 cells that are required for the pathogenesis of colitis. Natl. Acad. Sci. USA 2015, 112, 7061–7066. https://doi.org/10.1073/pnas.1415675112.
- Poholek, C.; Dulson, S.J.; Zajac, A.J.; Harrington, L.E. IL-21 Controls ILC3 Cytokine Production and Promotes a Protective Phenotype in a Mouse Model of Colitis. ImmunoHorizons 2019, 3, 194–202. https://doi.org/10.4049/immunohorizons.1900005.
- Kole, A.; He, J.; Rivollier, A.; Silveira, D.D.; Kitamura, K.; Maloy, K.J.; Kelsall, B.L. Type I IFNs Regulate Effector and Regulatory T Cell Accumulation and Anti-Inflammatory Cytokine Production during T Cell–Mediated Colitis. Immunol. 2013, 191, 2771–2779. https://doi.org/10.4049/jimmunol.1301093.
- Fort, M.M.; Leach, M.W.; Rennick, D.M. A role for NK cells as regulators of CD4+ T cells in a transfer model of colitis. Immunol. 1998, 161, 3256–3261.
- A Keilbaugh, S.; E Shin, M.; Banchereau, R.F.; McVay, L.D.; Boyko, N.; Artis, D.; Cebra, J.J.; Wu, G.D. Activation of RegIII / and interferon expression in the intestinal tract of SCID mice: An innate response to bacterial colonisation of the gut. Gut 2005, 54, 623–629. https://doi.org/10.1136/gut.2004.056028.
- Kjellev, S.; Haase, C.; Lundsgaard, D.; Ursø, B.; Tornehave, D.; Markholst, H. Inhibition of NKG2D receptor function by antibody therapy attenuates transfer-induced colitis in SCID mice. J. Immunol. 2007, 37, 1397–1406. https://doi.org/10.1002/eji.200636473.
- Yusung, S.; McGovern, D.; Lin, L.; Hommes, D.; Lagishetty, V.; Braun, J. NK cells are biologic and biochemical targets of 6-mercaptopurine in Crohn’s disease patients. Immunol. 2017, 175, 82–90. https://doi.org/10.1016/j.clim.2016.12.004.
- Bosma, G.C.; Fried, M.; Custer, R.P.; Carroll, A.; Gibson, D.M.; Bosma, M.J. Evidence of functional lymphocytes in some (leaky) scid mice. Exp. Med. 1988, 167, 1016–1033. https://doi.org/10.1084/jem.167.3.1016.
- Karo, J.M.; Schatz, D.G.; Sun, J.C. The RAG Recombinase Dictates Functional Heterogeneity and Cellular Fitness in Natural Killer Cells. Cell 2014, 159, 94–107. https://doi.org/10.1016/j.cell.2014.08.026.
- Kwan, A.; Abraham, R.S.; Currier, R.; Brower, A.; Andruszewski, K.; Abbott, J.; Baker, M.; Ballow, M.; Bartoshesky, L.E.; Bonilla, F.A.; et al. Newborn Screening for Severe Combined Immunodeficiency in 11 Screening Programs in the United States. JAMA 2014, 312, 729–738. https://doi.org/10.1001/jama.2014.9132.
- Mestas, J.; Hughes, C.C.W. Of mice and not men: Differences between mouse and human immunology. Immunol. 2004, 172, 2731–2738. https://doi.org/10.4049/jimmunol.172.5.2731.
- Chia, R.; Achilli, F.; Festing, M.F.W.; Fisher, E.M.C. The origins and uses of mouse outbred stocks. Genet. 2005, 37, 1181–1186. https://doi.org/10.1038/ng1665.