Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Zheng, L.; Pang, Q.; Xu, H.; Guo, H.; Liu, R.; Wang, T. Neurobiological Links between Stress and Traumatic Brain Injury. Encyclopedia. Available online: https://encyclopedia.pub/entry/26902 (accessed on 08 August 2026).
Zheng L, Pang Q, Xu H, Guo H, Liu R, Wang T. Neurobiological Links between Stress and Traumatic Brain Injury. Encyclopedia. Available at: https://encyclopedia.pub/entry/26902. Accessed August 08, 2026.
Zheng, Lexin, Qiuyu Pang, Heng Xu, Hanmu Guo, Rong Liu, Tao Wang. "Neurobiological Links between Stress and Traumatic Brain Injury" Encyclopedia, https://encyclopedia.pub/entry/26902 (accessed August 08, 2026).
Zheng, L., Pang, Q., Xu, H., Guo, H., Liu, R., & Wang, T. (2022, September 06). Neurobiological Links between Stress and Traumatic Brain Injury. In Encyclopedia. https://encyclopedia.pub/entry/26902
Zheng, Lexin, et al. "Neurobiological Links between Stress and Traumatic Brain Injury." Encyclopedia. Web. 06 September, 2022.
Copy Citation
Neurological dysfunctions commonly occurs after mild or moderate traumatic brain injury (TBI). Although most TBI patients recover from such dysfunction in a short period of time, some present with persistent neurological deficits. Stress is a potential factor that is involved in recovery from neurological dysfunction after TBI. However, there has been limited research on the effects and mechanisms of stress on neurological dysfunctions due to TBI. The effects of TBI and stress on neurological dysfunctions and different brain regions such as the prefrontal cortex, hippocampus, amygdala, and hypothalamus are investigated, and the neurobiological links and mechanisms between stress and TBI are explored.
traumatic brain injury
stress
brain region
neurological dysfunction
biomarker
1. Introduction
More than 50 million people worldwide suffer from traumatic brain injury (TBI) each year and approximately 80% of people will experience one or more mild or moderate TBI event in their lifetime [1][2]. In China, the population-based mortality of TBI is approximately 13 cases per 100,000 people, which is similar to the rates reported in other countries [3]. Although most mild and moderate TBI patients recover in a short period, many experiences persistent neurological dysfunction [4][5]. Although the factors that lead to prolonged neurological symptoms after TBI remain unclear, a history of stress is one key factor that can affect the degree of neurological impairments [4][6]. Stress has diverse effects on various brain regions, including the prefrontal cortex (PFC), hippocampus and amygdala [7][8]. Thus, the adverse consequences of acute and chronic stressors on TBI are worth investigating. The hypothalamic–pituitary–adrenal (HPA) axis and the locus coeruleus–norepinephrine (LC-NE) system also play key roles in stress processing [9], and a further study of these pathways may help to identify relevant stress biomarkers.
In this entry, the neurobiological links between neurological dysfunctions and the key brain regions affected by TBI and stress are first reviewed. Next, the effects and mechanisms of stress on neurological dysfunctions after TBI are discussed. Lastly, researchers aim to identify suitable stress markers and explore the possible diagnostic and therapeutic significance on stress or stress plus mild or moderate TBI.
2. Neurobiological Links between TBI and Neurological Dysfunctions
TBI is defined as an alteration in brain structure, or other evidence of brain pathology, caused by an external force [10][11]. From the aspect of distribution of structural damage, researchers can classify the injury to focal or diffuse [12]. Focal brain injury is caused by the outside forces acting on the skull and resulting in compression of the tissue underneath the cranium at the site of the impact or the tissue opposite to the impact [13]. The location of the impact to the skull determines the cerebral pathology and neurological deficits. By definition, diffuse brain injury is more scattered, and is not linked to a specific focus of destructive tissue damage [14], including widely distributed damage to axons, diffuse vascular injury, hypoxic–ischemic injury, and brain swelling. Regardless of focal or diffuse injury, the characteristic of TBI involves the mechanisms of primary and secondary brain injury [15]. Primary brain injury occurs at the exact moment of insult and results in the disruption of cell plasma membrane [16]. Secondary brain injury occurs after primary brain injury and involves the participation of complicated mechanisms, including excitatory toxicity, mitochondrial dysfunction, oxidative stress, lipid peroxidation, neuroinflammation, and axonal degeneration, and, finally, it induces diverse forms of programmed cell death, such as necroptosis, autophagy, apoptosis, pyroptosis, and ferroptosis [17][18][19][20][21]. TBI induces an increased mitochondrial membrane permeability. Mitochondria trigger a variety of apoptotic signaling pathways via interactions among the bcl-2 family proteins in order to release pro-apoptotic proteins from the intermembrane of mitochondria, which result in apoptosis [22]. Several events following TBI, such as tumor necrosis factor (TNF) release, toll-like receptors (TLR) activation, inflammation, and reactive oxygen species (ROS) production, have the potential to activate necrosis, which involves the upstream assembly of the necroptosome complex formed by the interaction of receptor interacting protein kinase 1 and 3 (RIPK1 and 3) and the downstream RIPK3-mediated phosphorylation of mixed lineage kinase domain-like (MLKL) protein [23], which result in necroptosis. Far more than that, the extent of cell loss following TBI has been correlated with cognitive deficits and long-term prognosis in both clinical and experimental studies [24].
A correct assessment of the degree of injury is essential for the effective treatment of TBI. TBI severity is traditionally determined by several clinical indicators, including the consciousness state, Glasgow coma scale score, presence/duration of retrograde amnesia, and neuroimaging evidence [25][26]. Neurological dysfunction can be divided into physical symptoms and neuropsychiatric symptoms. Neuropsychiatric dysfunctions, including cognitive impairments (executive dysfunction, attention disorder, or memory problems) and emotional/behavioral disorders (depression, anxiety, or sleep disorders) [11][27] are common after TBI, typically lasting 7 to 10 days but sometimes months to years. Therefore, differences in neurological dysfunctions after TBI depend on the severity of brain injury as well as the characteristics of the underlying key brain regions (Figure 1).
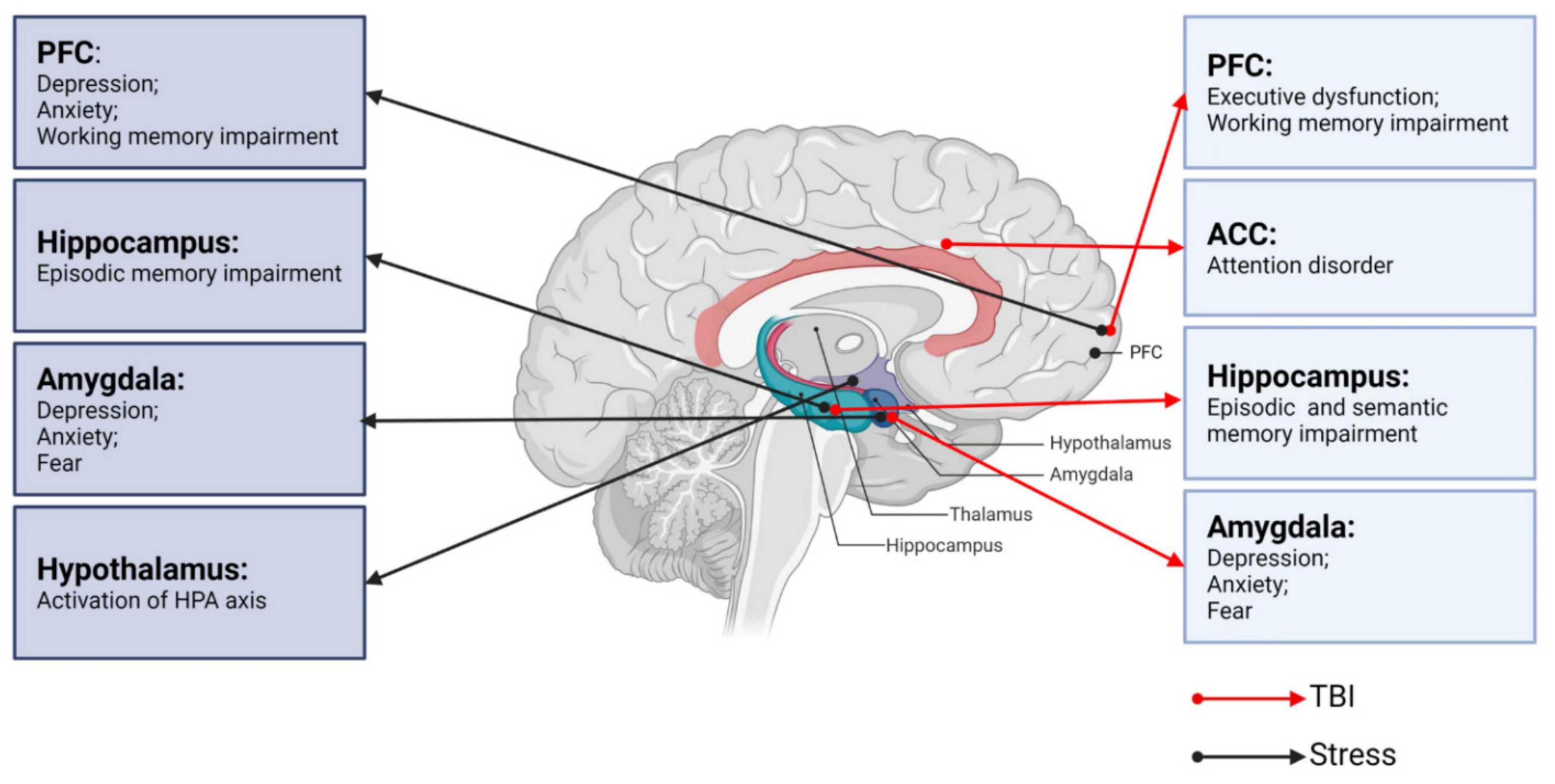
Figure 1. Neurobiological links between TBI, stress, and neurological dysfunctions. PFC: prefrontal cortex; ACC: anterior cingulate cortex; TBI: traumatic brain injury.
2.1. TBI and Cognitive Impairments
Forms of cognitive impairments post-TBI range from difficulties with executive function, i.e., attention and problem solving, to deficits in information processing and short- and long-term memory [28][29]. Previous research has shown that the cognitive functions, including memory, attention, and executive function, are resolved within 3 to 6 months after mild or moderate TBI [30], whereas severe TBI can cause cognitive impairments for 6 months or longer [28].
Funahashi [31] describes executive function as a product of the coordinated operation of various processes to accomplish a particular goal in a flexible manner. Executive function is the province of the PFC, and executive dysfunction post-TBI is typically associated with frontal lobe injury. Executive function is mediated by a distributed network guided by the frontal lobe that includes the prefrontal sub-region, posterior cortex, and subcortical structures, such as the basal ganglia and ventral striatum [32]. TBI causes the frontal lobe and subcortical structures, such as the cingulate gyrus, amygdala, striatum, and insula, to be particularly vulnerable [33]. Accordingly, patients with PFC damage show alterations in judgment, organization, planning, and decision making. There are functional and anatomical links between the frontal cortex and striatum [34]. Executive function relies on the efficient operation of cortical striatum circuits, which are often abnormal in cases of executive dysfunction [35][36]. The striatum can be divided into the caudate nucleus, putamen, and nucleus accumbens (NAc) [37]. The disruption of the striatum has been reported in various disorders involving executive dysfunction, such as Huntington’s disease [38], multiple system atrophy (MSA) [39], progressive supranuclear palsy [40], and attention deficit hyperactivity disorder (ADHD) [41]. Working memory is also an important part of executive function. Significant functional changes in the PFC circuit have been shown to reduce the large-scale patterns of brain activity, which is associated with working memory impairment [42][43].
Attention is characterized as selectivity and intensity [44]. Selectivity includes focalized attention with the inhibition of distractors and divided attention, which allows for the performance of two tasks simultaneously [44]. Intensity consists of sustained attention allowing the person to maintain attention levels over prolonged periods of time, and to be kept alert [44]. Impaired attention is one of the most common complaints of TBI survivors [45][46][47]. Based on neuroimaging studies, some psychiatric symptoms have been shown to correspond to functional abnormalities in brain regions, such as selective attention being localized to the anterior cingulate cortex (ACC) [48]. An insufficient activation of ACC can lead to reduced attention to detail and easy distraction. The ACC regulates the activity of the cortex and subcortical regions and influences the ability to control and coordinate their interactions [49][50][51]. Interestingly, Sheth et al. [52] reported resting state functional hyperconnectivity of the ACC in veterans with mild TBI (mTBI).
Several theories about learning and memory propose a stable link between the hippocampus and cortex that strengthens or consolidates memory [53]. Memory impairments caused by TBI may thus be due to alterations in the physiological circuit involving the cortex and hippocampus. The division of memory into short-term and long-term memory remains controversial [54], and memory can be alternatively divided into working, episodic, and semantic memory. The hippocampus is the core of episodic memory and the disruption of the dentate gyrus (DG), CA1, and CA3 regions is thought to be the main cause of episode memory deficits post-TBI [42]. A prior study demonstrated a decreased hippocampal volume in mTBI patients with episodic memory impairment [55]. The integrity of the hippocampus is also important for semantic memory. For example, Klooster et al. [56] reported impoverished semantic memory in patients with hippocampal amnesia. Manns et al. [57] assessed the semantic memory capacity of patients with hippocampus damage and found that semantic memory abilities were impaired—especially anterograde and retrograde memory.
2.2. TBI and Emotional/Behavioral Disturbances
The most common chronic emotional/behavioral disturbances that occur after TBI are depression, anxiety, and fear [58]. The amygdala is a key brain region involved in emotional processing [59] and amygdala damage is associated with emotional disorders similar to those that arise after TBI [58][60]. The amygdala also plays an important role in recognizing facial emotion, such as fear, disgust, and anger [61][62].
Depression, which is one of the most common chronic psychoses after TBI [63], is thought to be closely related to changes in the amygdala. Depressed patients often have some negative symptoms, such as sleep disturbance, fatigue (anergia), difficulty with concentration, and anhedonia (apathy) [64]. Studies have shown that depression is associated with increased activation in marginal regions, through which, the amygdala is richly associated with cortical regions [65][66]. The effect of antidepressants is partly via effects on the coupling of the amygdala with other brain regions [67]. In a functional neuroimaging study of patients with diffuse TBI [68], the amygdala was found to process emotions and regulate behavioral and physiological responses to stressors [69]. The PFC, as a significant center of thinking and behavior regulation, is also associated with depression [70]. The PFC can be divided into medial PFC (mPFC) and dorsolateral PFC (dPFC) [71]. Using diffusion tensor tractography, Jang et al. identified dPFC after TBI accompanied by depressive symptoms [72]. The hippocampus is part of the limbic system and has nerve fiber connections with emotion-related brain regions such as the PFC and amygdala. A decrease in hippocampal volume has been observed in patients with TBI [73]. In addition, hippocampal volume is associated with TBI injury severity and neuropsychological function [74].
Anxiety is a common condition in which an anxious mood or state persists without an immediate threat. The amygdala, which plays a key role in regulating anxiety-related behaviors [69], is composed of several parts. Of these, the basolateral amygdala (BLA) and central amygdala (CeA) are particularly important in anxiety management [75][76]. The BLA consists of 80% pyramidal glutamate (Glu) neurons, and 20% γ-aminobutyric acid (GABA) neurons. The CeA, which encompasses the centrolateral (CeL) and centromedial (CeM) nuclei, consists of 95% GABAergic medium spiny neurons [77]. The CeM, which is the main output region of the amygdala, mediates the autonomic and behavioral responses to anxiety via projections to the brainstem [78]. The balance between excitation and inhibition determines the overall degree of amygdala excitability. The hypoactivity of GABAergic neurons and/or an increased activation of glutamatergic neurons leads to amygdala hyperexcitability that manifests as anxiety [79]. Prior research suggested that TBI-induced anxiety-like behaviors were associated with increased glutamatergic neurons and decreased GABAergic neurons within the amygdala [68]. Figueiredo et al. [69] observed that animals exhibited increased anxiety-like behaviors 30 days after TBI.
The amygdala is also key for the acquisition and storage of fearful memory [80]. The BLA is the main region associated with sensory inputs into the amygdala, while the CeM is known as the fear effector structure [81]. The amygdala is also involved in regulating fear-related learning via interactions with other brain regions, such as the cortex and hippocampus. External stimuli information is processed through mechanisms inherent in the amygdala and by interactions with other brain regions to produce fear responses as an output and regulate fear responses [82]. Similarly, Glu receptors and GABA receptors are essential for fear learning and memory [83]. A recent study [84] using single-nucleus RNA sequencing (snRNA-seq) demonstrated a significant increase in Decorin (a small leucinerich proteoglycans) expression in amygdala excitatory neurons after TBI, whereas the knockout of Decorin alleviated TBI-related fear conditioning.
Overall, immediate or secondary pathological changes following TBI can lead to abnormal cognitive and emotional function. Through clinical interventions, most non-severe TBI patients recover in a short period without any sequelae. However, some non-severe TBI patients still show delayed and even severe neurological abnormalities [85][86]. Previous studies have found that stress may aggravate or improve neurological dysfunctions following non-severe TBI [87][88].
3. Neurobiological Links between Stress and Key Brain Regions
All organisms maintain a complex dynamic equilibrium or homeostasis that is constantly challenged by internal or external stimuli, which are termed stressors [89]. Physiological stress is beneficial to the body in that the body can quickly adapt to changes in internal and external environmental factors. However, pathological stress that is intense and persistent is harmful to the body and can cause physical and mental dysfunction, resulting in many negative adaptation reactions [90]. The brain processes external information and determines the necessary behavior and physiological responses, whether adapting or overloading. As an organ, the brain changes in response to acute and chronic stress [91], and stress hormones can have protective or destructive effects on the brain. Studies have shown that moderate stress facilitated classical conditioning and associative learning [92], in contrast to the chronic stress-induced deficits in spatial and contextual memory and attention [93]. It is noteworthy that different stress paradigms can have a psychological influence or physical impact simultaneously. Psychological stressors may include social order conflicts and competition for resources, as well as restraint and immobilization with accompanying anxiety and fear [94]. Methods of physical stress include, but are not limited to, a lack of food or water, handling, and surgical procedures [95]. The HPA axis and LC-NE system play major roles in these stress responses. Neural circuits between different brain regions, including the hippocampus, amygdala, PFC, and hypothalamus, also play pivotal roles in responding to stress (Figure 1) [96].
3.1. Stress and the HPA Axis
The activation of the HPA axis is the primary hormonal response to stress. The initiation of the HPA axis is controlled by corticotropin-releasing hormone (CRH) neurons of the paraventricular nucleus (PVN) [97]. CRH and arginine vasopressin (AVP), which are released by PVN through the pituitary portal to the pituitary gland, act together, acting on the pituitary gland to promote the release of adrenocorticotropic hormone (ACTH) via the circulatory system to the adrenal cortex, thereby promoting the synthesis and release of glucocorticoids (GCs), which act on the body’s organ systems to adapt to changes in the internal and external environment [98]. GCs primarily bind to two receptors in the brain, namely the mineralocorticoid receptor (MR) and GC receptor (GR). The activation of these receptors alters the gene expression profiles slowly and persistently, ultimately affecting brain function [97][99]. A prior study showed that GCs are beneficial for short-term adaptation, but that long-term administration can cause severe damages [100].
3.2. Stress and the LC-NE System
The LC in the brainstem contains NE-synthesizing neurons that send diffuse projections throughout the central nervous system (CNS). The LC-NE system plays a major role in behavioral and autonomic responses to stress. During a stressful period, LC-NE neurons supply NE across the CNS to modulate the central stress response [101][102]. NE acts on different adrenal receptors (α1, α2, and β) and exerts a powerful neuroregulatory function. NE has a higher affinity for α2-adrenergic receptors and a lower affinity for α1- and β-adrenergic receptors [103]. The LC is involved in the stress response mainly through β receptors located in the BLA [104].
3.3. Stress and the PFC
The mPFC is primarily composed of glutamatergic pyramidal neurons (PNs) [105]. The mPFC PNs that orchestrate stress responses are tightly controlled by a complex network of GABAergic interneurons [106]. Although there are several groups of interneurons, the majority of GABAergic interneurons express parvalbumin (PV) and are thus termed PV neurons. Somatostatin (SST) neurons are also thought to regulate the Glu output from the dendritic trees of PNs through synaptic contact [107]. An imbalance between excitatory and inhibitory (E/I) neurotransmission is thought to be the basis for various neuropsychiatric disorders [108]. These interneurons express GRs and have the ability to integrate systemic stress signals. GRs are bound during acute stress, increasing soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) protein complexes (which mediate synaptic vesicles to fuse with the anterior membrane) in the presynaptic membrane [109][110]. Therefore, acute stress increases the excitability of glutamatergic neurons, as shown by extracellular Glu, postsynaptic membrane N-methyl d-aspartate receptor (NMDAR), and alpha-amino-3-hydroxy-5-methyl-4-isoxazole-propionic acid receptor (AMPAR) expressions, and increases in NMDA and AMPA-mediated excitatory currents. Animals studies have shown that acute exposure to stress or the administration of GCs increased the Glu release from the PFC [110][111]. Using microdialysis, it has been shown that exposing rats to tail-pinch, restraint, or forced-swim stress induces a marked, transient increase in extracellular Glu levels in the PFC [107]. Conversely, repeated stress has been shown to inhibit Glu delivery in the PFC by promoting the degradation of Glu receptors in juvenile rats [111].
The PFC has extensive neuronal connections with other brain regions that regulate behavior, cognition, and emotions [112]. Catecholaminergic neurons projections to the cerebral cortex stem from two main sources, namely NE neurons of the LC in the brainstem and dopamine (DA) neurons of the ventral tegmental area (VTA) in the midbrain. The PFC is a main cortical target of both NE and DA innervations [113]. NE and DA each have an ‘inverted U’-shaped influence on working memory, such that either too little or too much impairs PFC function [114][115]. A previous study showed that α1-adrenoceptor stimulation in the PFC contributes to stress-induced cognitive impairments [116]. The low NE level present under control (non-stress) conditions optimizes working memory by engaging α2A-receptors, whereas the high NE level during stress impairs PFC function by stimulating lower-affinity α1-receptors and β1-receptors [117]. An excessive activation or blocking of dopamine D1 receptors (D1Rs) during working memory stress can both lead to working memory deficits [114]. Memory deficits due to the excessive activation of D1R can be prevented by D1R antagonists [118], whereas spatial working memory deficits mediated by an increased D1R density are improved by D1R agonists [119]. Under normal circumstances, the extensive connections of PFC coordinate brain activity and regulate the catecholamine input.
3.4. Stress and Hippocampus
The hippocampus is rich in GRs and MRs [120], making it a key regulatory region of the HPA axis. Stress has a great impact on excitatory transmission and the synaptic plasticity of the hippocampus [121]. Excitatory amino acids and NMDAR play important roles in episodic memory function, which is dominated by the hippocampus. Excitatory amino acids produce long-term potentiation (LTP) on synapses [122]. Thus, the plasticity of synaptic connections in the hippocampus and LTP are the basis of learning and memory. LTP production relies on synaptic connections between cells in the CA1 and CA3 regions of the hippocampus, which, in turn, depend on Glu as a neurotransmitter [123]. During stress, GCs increase and stimulate Glu release from the hippocampus, which, in turn, inhibits DG proliferation [124]. Studies have shown that exposure to predator odor (2,4,5-trimethythiazole, TMT) causes a stress response in rats, as demonstrated by elevated adrenal steroid levels, DG excitation, and the rapid inhibition of DG proliferation [125]. CA3 dendritic atrophy is suppressed when an excitatory input pathway is damaged. Antagonism with NMDAR can inhibit stress-induced CA3 dendritic atrophy [126]. Interestingly, morphological damage of the CA3 region was reversed within 21 days in a rat model after the end of chronic stress [127]. Chronic stress can also produce CA1 apical dendritic retraction, although stressors tend to be more severe than what is needed to produce an apical dendritic retraction in the CA3 region [128].
3.5. Stress and Amygdala
The amygdala plays a key role in physiological and behavioral responses to stress and is characterized by high inhibitory tension mediated by GABA at rest. Stress causes hyperactivity of the amygdala, which is often accompanied by a reduction in inhibition controls [129]. Under physiological conditions, mPFC exerts top-down inhibitory control over amygdala activity, limiting its output and thereby preventing an inappropriate expression of emotions. Under stress conditions, the amygdala activates stress pathways in the hypothalamus and brainstem to induce high levels of NE and DA release, thereby impairing PFC regulation but strengthening amygdala function [130]. In such cases, the PFC control of stress becomes defective, resulting in aberrant amygdala activation and deficits in emotion and behavior [131]. Thus, during stress, the orchestration of the brain’s response patterns switches from the slow, thoughtful PFC regulation to the reflexive, rapid emotional regulation mediated by the amygdala and related subcortical structures [11].
Stress causes the remodeling of amygdala neuronal projections [132]. In contrast to stress-induced dendritic retraction seen in the hippocampus, projecting neurons within the BLA showed persistent dendritic hypertrophy after chronic stress, but dendritic contractions after acute stress [7]. Thus, the morphology of the amygdala and hippocampus showed opposite adjustments after chronic stress. In contrast, Glu is enhanced in both the amygdala and hippocampus after stress. The increase in Glu levels activates NMDAR in BLA, thereby delaying the increase in synaptic spines. A prior study showed that the GR agonist dexamethasone (DEX) enhanced fear resolution via a dose-dependent regulation of the methylation of the GR partner FK506-binding protein 5 (FKBP5) in the BLA [133]. The amygdala is also a major extrahypothalamic source of corticotropin releasing factor (CRF)-containing neurons and has high expression levels of the two cognate CRF receptors. During chronic stress, the repeated activation of CRF receptors leads to an increased NMDAR-mediated Ca2+ inflow [134], which inhibits the polymerization of tubulin dimers responsible for microtubule and neurite elongation. If Ca2+ is sustained at high levels, microtubules and microfilaments will be depolymerized to trigger dendritic regression [135].
Thus, under stress conditions, the HPA axis and LC-NE systems act with central GRs through the final metabolites GCs and NE. The activation of MRs and adrenal receptors affects the balance of excitatory and inhibitory neurons in the PFC, hippocampus, and amygdala, as well as neuronal plasticity, ultimately affecting working memory, emotions, and other neurological functions. Taken together, these findings indicate that both stress and TBI can lead to deficits in the corresponding brain regions.
References
- Maas, A.I.R.; Menon, D.K.; Adelson, P.D.; Andelic, N.; Bell, M.J.; Belli, A.; Bragge, P.; Brazinova, A.; Büki, A.; Chesnut, R.M.; et al. Traumatic brain injury: Integrated approaches to improve prevention, clinical care, and research. Lancet Neurol. 2017, 16, 987–1048.
- Feigin, V.L.; Theadom, A.; Barker-Collo, S.; Starkey, N.J.; McPherson, K.; Kahan, M.; Dowell, A.; Brown, P.; Parag, V.; Kydd, R.; et al. Incidence of traumatic brain injury in New Zealand: A population-based study. Lancet Neurol. 2013, 12, 53–64.
- Jiang, J.-Y.; Gao, G.-Y.; Feng, J.-F.; Mao, Q.; Chen, L.-G.; Yang, X.-F.; Liu, J.-F.; Wang, Y.-H.; Qiu, B.-H.; Huang, X.-J. Traumatic brain injury in China. Lancet Neurol. 2019, 18, 286–295.
- Howe, E.I.; Langlo, K.S.; Terjesen, H.C.A.; Røe, C.; Schanke, A.K.; Søberg, H.L.; Sveen, U.; Aas, E.; Enehaug, H.; Alves, D.E.; et al. Combined cognitive and vocational interventions after mild to moderate traumatic brain injury: Study protocol for a randomized controlled trial. Trials 2017, 18, 483.
- Markovic, S.J.; Fitzgerald, M.; Peiffer, J.J.; Scott, B.R.; Rainey-Smith, S.R.; Sohrabi, H.R.; Brown, B.M. The impact of exercise, sleep, and diet on neurocognitive recovery from mild traumatic brain injury in older adults: A narrative review. Ageing Res. Rev. 2021, 68, 101322.
- Fischer, J.T.; Bickart, K.C.; Giza, C.; Babikian, T. A Review of Family Environment and Neurobehavioral Outcomes Following Pediatric Traumatic Brain Injury: Implications of Early Adverse Experiences, Family Stress, and Limbic Development. Biol. Psychiatry 2022, 91, 488–497.
- Zhang, J.Y.; Liu, T.H.; He, Y.; Pan, H.Q.; Zhang, W.H.; Yin, X.P.; Tian, X.L.; Li, B.M.; Wang, X.D.; Holmes, A.; et al. Chronic Stress Remodels Synapses in an Amygdala Circuit-Specific Manner. Biol. Psychiatry 2019, 85, 189–201.
- Merino, E.; Raya-Salom, D.; Teruel-Martí, V.; Adell, A.; Cervera-Ferri, A.; Martínez-Ricós, J. Effects of Acute Stress on the Oscillatory Activity of the Hippocampus–Amygdala–Prefrontal Cortex Network. Neuroscience 2021, 476, 72–89.
- Kazakou, P.; Nicolaides, N.C.; Chrousos, G.P. Basic Concepts and Hormonal Regulators of the Stress System. Horm. Res. Paediatr. 2022.
- Menon, D.K.; Schwab, K.; Wright, D.W.; Maas, A.I. Demographics and Clinical Assessment Working Group of the International and Interagency Initiative toward Common Data Elements for Research on Traumatic Brain Injury and Psychological Health. Position statement: Definition of traumatic brain injury. Arch. Phys. Med. Rehabil. 2010, 91, 1637–1640.
- Pavlovic, D.; Pekic, S.; Stojanovic, M.; Popovic, V. Traumatic brain injury: Neuropathological, neurocognitive and neurobehavioral sequelae. Pituitary 2019, 22, 270–282.
- Andriessen, T.M.; Jacobs, B.; Vos, P.E. Clinical characteristics and pathophysiological mechanisms of focal and diffuse traumatic brain injury. J. Cell. Mol. Med. 2010, 14, 2381–2392.
- Pudenz, R.H.; Shelden, C.H. The Lucite Calvarium—A Method for Direct Observation of the Brain: II. Cranial Trauma and Brain Movement. J. Neurosurg. 1946, 3, 487–505.
- McGinn, M.J.; Povlishock, J.T. Pathophysiology of Traumatic Brain Injury. Neurosurg. Clin. N. Am. 2016, 27, 397–407.
- Stocchetti, N.; Carbonara, M.; Citerio, G.; Ercole, A.; Skrifvars, M.B.; Smielewski, P.; Zoerle, T.; Menon, D.K. Severe traumatic brain injury: Targeted management in the intensive care unit. Lancet Neurol. 2017, 16, 452–464.
- Pearn, M.L.; Niesman, I.R.; Egawa, J.; Sawada, A.; Almenar-Queralt, A.; Shah, S.B.; Duckworth, J.L.; Head, B.P. Pathophysiology Associated with Traumatic Brain Injury: Current Treatments and Potential Novel Therapeutics. Cell. Mol. Neurobiol. 2017, 37, 571–585.
- Wang, K.; Cui, D.M.; Gao, L. Traumatic brain injury: A review of characteristics, molecular basis and management. Front. Biosci. 2016, 21, 890–899.
- Sullivan, P.G.; Rabchevsky, A.G.; Waldmeier, P.C.; Springer, J.E. Mitochondrial permeability transition in CNS trauma: Cause or effect of neuronal cell death? J. Neurosci. Res. 2005, 79, 231–239.
- Khatri, N.; Thakur, M.; Pareek, V.; Kumar, S.; Sharma, S.; Datusalia, K.A. Oxidative Stress: Major Threat in Traumatic Brain Injury. CNS Neurol. Disord. Drug Targets 2018, 17, 689–695.
- Anthonymuthu, T.S.; Kenny, E.M.; Bayır, H. Therapies targeting lipid peroxidation in traumatic brain injury. Brain Res. 2016, 1640, 57–76.
- Ladak, A.A.; Enam, S.A.; Ibrahim, M.T. A Review of the Molecular Mechanisms of Traumatic Brain Injury. World Neurosurg. 2019, 131, 126–132.
- Akamatsu, Y.; Hanafy, K.A. Cell Death and Recovery in Traumatic Brain Injury. Neurotherapeutics 2020, 17, 446–456.
- Wehn, A.C.; Khalin, I.; Duering, M.; Hellal, F.; Culmsee, C.; Vandenabeele, P.; Plesnila, N.; Terpolilli, N.A. RIPK1 or RIPK3 deletion prevents progressive neuronal cell death and improves memory function after traumatic brain injury. Acta Neuropathol. Commun. 2021, 9, 138.
- Walker, K.R.; Tesco, G. Molecular mechanisms of cognitive dysfunction following traumatic brain injury. Front. Aging Neurosci. 2013, 5, 29.
- Teasdale, G.; Jennett, B. Assessment of Coma and Impaired Consciousness. Lancet 1974, 304, 81–84.
- Friedland, D.; Swash, M. Post-traumatic amnesia and confusional state: Hazards of retrospective assessment. J. Neurol. Neurosurg. Psychiatry 2016, 87, 1068–1074.
- Rao, V.; Syeda, A.; Roy, D.; Peters, M.E.; Vaishnavi, S. Neuropsychiatric aspects of concussion: Acute and chronic sequelae. Concussion 2017, 2, CNC29.
- Tucker, L.B.; Velosky, A.G.; McCabe, J.T. Applications of the Morris water maze in translational traumatic brain injury research. Neurosci. Biobehav. Rev. 2018, 88, 187–200.
- Stocchetti, N.; Zanier, E.R. Chronic impact of traumatic brain injury on outcome and quality of life: A narrative review. Crit. Care 2016, 20, 148.
- Rabinowitz, A.R.; Levin, H.S. Cognitive sequelae of traumatic brain injury. Psychiatr. Clin. N. Am. 2014, 37, 1–11.
- Funahashi, S. Neuronal mechanisms of executive control by the prefrontal cortex. Neurosci. Res. 2001, 39, 147–165.
- Levin, H.S.; Hanten, G. Executive functions after traumatic brain injury in children. Pediatr. Neurol. 2005, 33, 79–93.
- Wood, R.L.; Worthington, A. Neurobehavioral Abnormalities Associated with Executive Dysfunction after Traumatic Brain Injury. Front. Behav. Neurosci. 2017, 11, 195.
- Alexander, G.E.; DeLong, M.R.; Strick, P.L. Parallel organization of functionally segregated circuits linking basal ganglia and cortex. Annu. Rev. Neurosci. 1986, 9, 357–381.
- De Simoni, S.; Jenkins, P.O.; Bourke, N.J.; Fleminger, J.J.; Hellyer, P.J.; Jolly, A.E.; Patel, M.C.; Cole, J.H.; Leech, R.; Sharp, D.J. Altered caudate connectivity is associated with executive dysfunction after traumatic brain injury. Brain 2018, 141, 148–164.
- Bamford, I.J.; Bamford, N.S. The Striatum’s Role in Executing Rational and Irrational Economic Behaviors. Neuroscientist 2019, 25, 475–490.
- Ketchesin, K.D.; Zong, W.; Hildebrand, M.A.; Seney, M.L.; Cahill, K.M.; Scott, M.R.; Shankar, V.G.; Glausier, J.R.; Lewis, D.A.; Tseng, G.C.; et al. Diurnal rhythms across the human dorsal and ventral striatum. Proc. Natl. Acad. Sci. USA 2021, 118, e2016150118.
- McColgan, P.; Seunarine, K.K.; Razi, A.; Cole, J.H.; Gregory, S.; Durr, A.; Roos, R.A.; Stout, J.C.; Landwehrmeyer, B.; Scahill, R.I.; et al. Selective vulnerability of Rich Club brain regions is an organizational principle of structural connectivity loss in Huntington’s disease. Brain 2015, 138, 3327–3344.
- Ndayisaba, A.; Jellinger, K.; Berger, T.; Wenning, G.K. TNFα inhibitors as targets for protective therapies in MSA: A viewpoint. J. Neuroinflamm. 2019, 16, 80.
- Elliott, R. Executive functions and their disorders: Imaging in clinical neuroscience. Br. Med. Bull. 2003, 65, 49–59.
- Mennes, M.; Vega Potler, N.; Kelly, C.; Di Martino, A.; Castellanos, F.X.; Milham, M.P. Resting state functional connectivity correlates of inhibitory control in children with attention-deficit/hyperactivity disorder. Front. Psychiatry 2011, 2, 83.
- Paterno, R.; Folweiler, K.A.; Cohen, A.S. Pathophysiology and Treatment of Memory Dysfunction after Traumatic Brain Injury. Curr. Neurol. Neurosci. Rep. 2017, 17, 52.
- Vakil, E.; Greenstein, Y.; Weiss, I.; Shtein, S. The Effects of Moderate-to-Severe Traumatic Brain Injury on Episodic Memory: A Meta-Analysis. Neuropsychol. Rev. 2019, 29, 270–287.
- Le Fur, C.; Câmara-Costa, H.; Francillette, L.; Opatowski, M.; Toure, H.; Brugel, D.; Laurent-Vannier, A.; Meyer, P.; Watier, L.; Dellatolas, G.; et al. Executive functions and attention 7years after severe childhood traumatic brain injury: Results of the Traumatisme Grave de l’Enfant (TGE) cohort. Ann. Phys. Rehabil. Med. 2020, 63, 270–279.
- Ornstein, T.J.; Sagar, S.; Schachar, R.J.; Ewing-Cobbs, L.; Chapman, S.B.; Dennis, M.; Saunders, A.E.; Yang, T.T.; Levin, H.S.; Max, J.E. Neuropsychological performance of youth with secondary attention-deficit/hyperactivity disorder 6- and 12-months after traumatic brain injury. J. Int. Neuropsychol. Soc. 2014, 20, 971–981.
- Anderson, V.; Eren, S.; Dob, R.; Le Brocque, R.; Iselin, G.; Davern, T.J.; McKinlay, L.; Kenardy, J. Early Attention Impairment and Recovery Profiles After Childhood Traumatic Brain Injury. J. Head Trauma Rehabil. 2012, 27, 199–209.
- Shah, S.A.; Goldin, Y.; Conte, M.M.; Goldfine, A.M.; Mohamadpour, M.; Fidali, B.C.; Cicerone, K.; Schiff, N.D. Executive attention deficits after traumatic brain injury reflect impaired recruitment of resources. NeuroImage Clin. 2017, 14, 233–241.
- Cazalis, F.; Babikian, T.; Giza, C.; Copeland, S.; Hovda, D.; Asarnow, R.F. Pivotal role of anterior cingulate cortex in working memory after traumatic brain injury in youth. Front. Neurol. 2011, 1, 158.
- Xuan, B.; Mackie, M.A.; Spagna, A.; Wu, T.; Tian, Y.; Hof, P.R.; Fan, J. The activation of interactive attentional networks. Neuroimage 2016, 129, 308–319.
- Bush, G.; Luu, P.; Posner, M.I. Cognitive and emotional influences in anterior cingulate cortex. Trends Cogn. Sci. 2000, 4, 215–222.
- Schneider, K.N.; Sciarillo, X.A.; Nudelman, J.L.; Cheer, J.F.; Roesch, M.R. Anterior Cingulate Cortex Signals Attention in a Social Paradigm that Manipulates Reward and Shock. Curr. Biol. 2020, 30, 3724–3735.e2.
- Sheth, C.; Rogowska, J.; Legarreta, M.; McGlade, E.; Yurgelun-Todd, D. Functional connectivity of the anterior cingulate cortex in Veterans with mild traumatic brain injury. Behav. Brain Res. 2021, 396, 112882.
- Whiting, M.D.; Hamm, R.J. Mechanisms of anterograde and retrograde memory impairment following experimental traumatic brain injury. Brain Res. 2008, 1213, 69–77.
- Cowan, N. Chapter 20 What are the differences between long-term, short-term, and working memory? In Essence of Memory; Elsevier: Amsterdam, The Netherlands, 2008; pp. 323–338.
- Fortier-Lebel, O.; Jobin, B.; Lecuyer-Giguere, F.; Gaubert, M.; Giguere, J.F.; Gagnon, J.F.; Boller, B.; Frasnelli, J. Verbal Episodic Memory Alterations and Hippocampal Atrophy in Acute Mild Traumatic Brain Injury. J. Neurotrauma 2021, 38, 1506–1514.
- Klooster, N.B.; Tranel, D.; Duff, M.C. The hippocampus and semantic memory over time. Brain Lang. 2020, 201, 104711.
- Manns, J.R.; Hopkins, R.O.; Squire, L.R. Semantic Memory and the Human Hippocampus. Neuron 2003, 38, 127–133.
- Van der Horn, H.J.; Liemburg, E.J.; Scheenen, M.E.; de Koning, M.E.; Marsman, J.B.; Spikman, J.M.; van der Naalt, J. Brain network dysregulation, emotion, and complaints after mild traumatic brain injury. Hum. Brain Mapp. 2016, 37, 1645–1654.
- Simic, G.; Tkalcic, M.; Vukic, V.; Mulc, D.; Spanic, E.; Sagud, M.; Olucha-Bordonau, F.E.; Vuksic, M.; Hof, P.R. Understanding Emotions: Origins and Roles of the Amygdala. Biomolecules 2021, 11, 823.
- Han, K.; Chapman, S.B.; Krawczyk, D.C. Altered Amygdala Connectivity in Individuals with Chronic Traumatic Brain Injury and Comorbid Depressive Symptoms. Front. Neurol. 2015, 6, 231.
- Young, L.R.; Yu, W.; Holloway, M.; Rodgers, B.N.; Chapman, S.B.; Krawczyk, D.C. Amygdala activation as a marker for selective attention toward neutral faces in a chronic traumatic brain injury population. Neuropsychologia 2017, 104, 214–222.
- Celeghin, A.; Galetto, V.; Tamietto, M.; Zettin, M. Emotion Recognition in Low-Spatial Frequencies Is Partly Preserved following Traumatic Brain Injury. BioMed Res. Int. 2019, 2019, 9562935.
- Lim, S.W.; Shiue, Y.L.; Liao, J.C.; Wee, H.Y.; Wang, C.C.; Chio, C.C.; Chang, C.H.; Hu, C.Y.; Kuo, J.R. Simvastatin Therapy in the Acute Stage of Traumatic Brain Injury Attenuates Brain Trauma-Induced Depression-Like Behavior in Rats by Reducing Neuroinflammation in the Hippocampus. Neurocrit. Care 2017, 26, 122–132.
- Silver, J.M.; McAllister, T.W.; Arciniegas, D.B. Depression and Cognitive Complaints Following Mild Traumatic Brain Injury. Am. J. Psychiatry 2009, 166, 653–661.
- Anand, A.; Li, Y.; Wang, Y.; Wu, J.; Gao, S.; Bukhari, L.; Mathews, V.P.; Kalnin, A.; Lowe, M.J. Activity and connectivity of brain mood regulating circuit in depression: A functional magnetic resonance study. Biol. Psychiatry 2005, 57, 1079–1088.
- Matthews, S.C.; Strigo, I.A.; Simmons, A.N.; Yang, T.T.; Paulus, M.P. Decreased functional coupling of the amygdala and supragenual cingulate is related to increased depression in unmedicated individuals with current major depressive disorder. J. Affect. Disord. 2008, 111, 13–20.
- Chen, C.H.; Suckling, J.; Ooi, C.; Fu, C.H.; Williams, S.C.; Walsh, N.D.; Mitterschiffthaler, M.T.; Pich, E.M.; Bullmore, E. Functional coupling of the amygdala in depressed patients treated with antidepressant medication. Neuropsychopharmacology 2008, 33, 1909–1918.
- Beitchman, J.A.; Griffiths, D.R.; Hur, Y.; Ogle, S.B.; Bromberg, C.E.; Morrison, H.W.; Lifshitz, J.; Adelson, P.D.; Thomas, T.C. Experimental Traumatic Brain Injury Induces Chronic Glutamatergic Dysfunction in Amygdala Circuitry Known to Regulate Anxiety-Like Behavior. Front. Neurosci. 2019, 13, 1434.
- Figueiredo, T.H.; Harbert, C.L.; Pidoplichko, V.; Almeida-Suhett, C.P.; Pan, H.; Rossetti, K.; Braga, M.F.M.; Marini, A.M. Alpha-Linolenic Acid Treatment Reduces the Contusion and Prevents the Development of Anxiety-Like Behavior Induced by a Mild Traumatic Brain Injury in Rats. Mol. Neurobiol. 2018, 55, 187–200.
- Treadway, M.T.; Waskom, M.L.; Dillon, D.G.; Holmes, A.J.; Park, M.T.M.; Chakravarty, M.M.; Dutra, S.J.; Polli, F.E.; Iosifescu, D.V.; Fava, M.; et al. Illness progression, recent stress, and morphometry of hippocampal subfields and medial prefrontal cortex in major depression. Biol. Psychiatry 2015, 77, 285–294.
- Liu, W.; Ge, T.; Leng, Y.; Pan, Z.; Fan, J.; Yang, W.; Cui, R. The Role of Neural Plasticity in Depression: From Hippocampus to Prefrontal Cortex. Neural Plast. 2017, 2017, 6871089.
- Jang, S.H.; Yi, J.H.; Kwon, H.G. Injury of the dorsolateral prefronto-thalamic tract in a patient with depression following mild traumatic brain injury: A case report. Medicine 2016, 95, e5009.
- Palacios, E.M.; Sala-Llonch, R.; Junque, C.; Fernandez-Espejo, D.; Roig, T.; Tormos, J.M.; Bargallo, N.; Vendrell, P. Long-term declarative memory deficits in diffuse TBI: Correlations with cortical thickness, white matter integrity and hippocampal volume. Cortex 2013, 49, 646–657.
- Bae, S.; Sheth, C.; Legarreta, M.; McGlade, E.; Lyoo, I.K.; Yurgelun-Todd, D.A. Volume and shape analysis of the Hippocampus and amygdala in veterans with traumatic brain injury and posttraumatic stress disorder. Brain Imaging Behav. 2020, 14, 1850–1864.
- Janak, P.H.; Tye, K.M. From circuits to behaviour in the amygdala. Nature 2015, 517, 284–292.
- Babaev, O.; Piletti Chatain, C.; Krueger-Burg, D. Inhibition in the amygdala anxiety circuitry. Exp. Mol. Med. 2018, 50, 1–16.
- Orsini, C.A.; Maren, S. Neural and cellular mechanisms of fear and extinction memory formation. Neurosci. Biobehav. Rev. 2012, 36, 1773–1802.
- Tye, K.M.; Prakash, R.; Kim, S.Y.; Fenno, L.E.; Grosenick, L.; Zarabi, H.; Thompson, K.R.; Gradinaru, V.; Ramakrishnan, C.; Deisseroth, K. Amygdala circuitry mediating reversible and bidirectional control of anxiety. Nature 2011, 471, 358–362.
- Agoglia, A.E.; Herman, M.A. The center of the emotional universe: Alcohol, stress, and CRF1 amygdala circuitry. Alcohol 2018, 72, 61–73.
- LeDoux, J.E. Emotion circuits in the brain. Annu. Rev. Neurosci. 2000, 23, 155–184.
- Duvarci, S.; Pare, D. Amygdala microcircuits controlling learned fear. Neuron 2014, 82, 966–980.
- Ehrlich, I.; Humeau, Y.; Grenier, F.; Ciocchi, S.; Herry, C.; Luthi, A. Amygdala inhibitory circuits and the control of fear memory. Neuron 2009, 62, 757–771.
- Makkar, S.R.; Zhang, S.Q.; Cranney, J. Behavioral and neural analysis of GABA in the acquisition, consolidation, reconsolidation, and extinction of fear memory. Neuropsychopharmacology 2010, 35, 1625–1652.
- Shi, Y.; Wu, X.; Zhou, J.; Cui, W.; Wang, J.; Hu, Q.; Zhang, S.; Han, L.; Zhou, M.; Luo, J.; et al. Single-Nucleus RNA Sequencing Reveals that Decorin Expression in the Amygdala Regulates Perineuronal Nets Expression and Fear Conditioning Response after Traumatic Brain Injury. Adv. Sci. 2022, 9, e2104112.
- Ramlackhansingh, A.F.; Brooks, D.J.; Greenwood, R.J.; Bose, S.K.; Turkheimer, F.E.; Kinnunen, K.M.; Gentleman, S.; Heckemann, R.A.; Gunanayagam, K.; Gelosa, G.; et al. Inflammation after trauma: Microglial activation and traumatic brain injury. Ann. Neurol. 2011, 70, 374–383.
- Johnson, V.E.; Stewart, J.E.; Begbie, F.D.; Trojanowski, J.Q.; Smith, D.H.; Stewart, W. Inflammation and white matter degeneration persist for years after a single traumatic brain injury. Brain 2013, 136, 28–42.
- The National Academies of Sciences, Engineering, and Medicine. Evaluation of the Disability Determination Process for Traumatic Brain Injury in Veterans; The National Academies Press: Washington, DC, USA, 2019; p. 210.
- Bay, E.; de-Leon, M.B. Chronic Stress and Fatigue-Related Quality of Life After Mild to Moderate Traumatic Brain Injury. J. Head Trauma Rehabil. 2011, 26, 355–363.
- Chrousos, G.P. Stress and disorders of the stress system. Nat. Rev. Endocrinol. 2009, 5, 374–381.
- Dragoş, D.; Tănăsescu, M.D. The effect of stress on the defense systems. J. Med. Life 2010, 3, 10–18.
- McEwen, B.S.; Gianaros, P.J. Stress- and allostasis-induced brain plasticity. Annu. Rev. Med. 2011, 62, 431–445.
- Joëls, M.; Pu, Z.; Wiegert, O.; Oitzl, M.S.; Krugers, H.J. Learning under stress: How does it work? Trends Cogn. Sci. 2006, 10, 152–158.
- Liston, C.; Miller, M.M.; Goldwater, D.S.; Radley, J.J.; Rocher, A.B.; Hof, P.R.; Morrison, J.H.; McEwen, B.S. Stress-induced alterations in prefrontal cortical dendritic morphology predict selective impairments in perceptual attentional set-shifting. J. Neurosci. 2006, 26, 7870–7874.
- Gameiro, G.H.; Gameiro, P.H.; da Silva Andrade, A.; Pereira, L.F.; Arthuri, M.T.; Marcondes, F.K.; de Arruda Veiga, M.C.F. Nociception- and anxiety-like behavior in rats submitted to different periods of restraint stress. Physiol. Behav. 2006, 87, 643–649.
- Buynitsky, T.; Mostofsky, D.I. Restraint stress in biobehavioral research: Recent developments. Neurosci. Biobehav. Rev. 2009, 33, 1089–1098.
- McEwen, B.S.; Gianaros, P.J. Central role of the brain in stress and adaptation: Links to socioeconomic status, health, and disease. Ann. N. Y. Acad. Sci. 2010, 1186, 190–222.
- Herman, J.P.; McKlveen, J.M.; Ghosal, S.; Kopp, B.; Wulsin, A.; Makinson, R.; Scheimann, J.; Myers, B. Regulation of the Hypothalamic-Pituitary-Adrenocortical Stress Response. Compr. Physiol. 2016, 6, 603–621.
- Herman, J.P.; Tasker, J.G. Paraventricular Hypothalamic Mechanisms of Chronic Stress Adaptation. Front. Endocrinol. 2016, 7, 137.
- Joels, M. Corticosteroids and the brain. J. Endocrinol. 2018, 238, R121–R130.
- Jankord, R.; Herman, J.P. Limbic regulation of hypothalamo-pituitary-adrenocortical function during acute and chronic stress. Ann. N. Y. Acad. Sci. 2008, 1148, 64–73.
- Ding, X.F.; Zhao, X.H.; Tao, Y.; Zhong, W.C.; Fan, Q.; Diao, J.X.; Liu, Y.L.; Chen, Y.Y.; Chen, J.X.; Lv, Z.P. Xiao Yao San Improves Depressive-Like Behaviors in Rats with Chronic Immobilization Stress through Modulation of Locus Coeruleus-Norepinephrine System. Evid. Based Complement. Altern. Med. 2014, 2014, 605914.
- Schwarz, L.A.; Luo, L. Organization of the locus coeruleus-norepinephrine system. Curr. Biol. 2015, 25, R1051–R1056.
- Arnsten, A.F. Through the looking glass: Differential noradenergic modulation of prefrontal cortical function. Neural Plast. 2000, 7, 133–146.
- Benarroch, E.E. Locus coeruleus. Cell Tissue Res. 2018, 373, 221–232.
- Harris, K.D.; Shepherd, G.M.G. The neocortical circuit: Themes and variations. Nat. Neurosci. 2015, 18, 170–181.
- McKlveen, J.M.; Moloney, R.D.; Scheimann, J.R.; Myers, B.; Herman, J.P. “Braking” the Prefrontal Cortex: The Role of Glucocorticoids and Interneurons in Stress Adaptation and Pathology. Biol. Psychiatry 2019, 86, 669–681.
- McKlveen, J.M.; Myers, B.; Herman, J.P. The medial prefrontal cortex: Coordinator of autonomic, neuroendocrine and behavioural responses to stress. J. Neuroendocrinol. 2015, 27, 446–456.
- McKlveen, J.M.; Morano, R.L.; Fitzgerald, M.; Zoubovsky, S.; Cassella, S.N.; Scheimann, J.R.; Ghosal, S.; Mahbod, P.; Packard, B.A.; Myers, B.; et al. Chronic Stress Increases Prefrontal Inhibition: A Mechanism for Stress-Induced Prefrontal Dysfunction. Biol. Psychiatry 2016, 80, 754–764.
- Popoli, M.; Yan, Z.; McEwen, B.S.; Sanacora, G. The stressed synapse: The impact of stress and glucocorticoids on glutamate transmission. Nat. Rev. Neurosci. 2011, 13, 22–37.
- Musazzi, L.; Milanese, M.; Farisello, P.; Zappettini, S.; Tardito, D.; Barbiero, V.S.; Bonifacino, T.; Mallei, A.; Baldelli, P.; Racagni, G.; et al. Acute stress increases depolarization-evoked glutamate release in the rat prefrontal/frontal cortex: The dampening action of antidepressants. PLoS ONE 2010, 5, e8566.
- Yuen, E.Y.; Wei, J.; Liu, W.; Zhong, P.; Li, X.; Yan, Z. Repeated stress causes cognitive impairment by suppressing glutamate receptor expression and function in prefrontal cortex. Neuron 2012, 73, 962–977.
- Segovia, G.; del Arco, A.; Mora, F. Environmental enrichment, prefrontal cortex, stress, and aging of the brain. J. Neural Transm. 2009, 116, 1007–1016.
- Xing, B.; Li, Y.-C.; Gao, W.-J. Norepinephrine versus dopamine and their interaction in modulating synaptic function in the prefrontal cortex. Brain Res. 2016, 1641, 217–233.
- Arnsten, A.F.T. Stress signalling pathways that impair prefrontal cortex structure and function. Nat. Rev. Neurosci. 2009, 10, 410–422.
- Aston-Jones, G.; Cohen, J.D. An integrative theory of locus coeruleus-norepinephrine function: Adaptive Gain and Optimal Performance. Annu. Rev. Neurosci. 2005, 28, 403–450.
- Birnbaum, S.; Gobeske, K.T.; Auerbach, J.; Taylor, J.R.; Arnsten, A.F.T. A role for norepinephrine in stress-induced cognitive deficits: α-1-adrenoceptor mediation in the prefrontal cortex. Biol. Psychiatry 1999, 46, 1266–1274.
- Ramos, B.P.; Arnsten, A.F.T. Adrenergic pharmacology and cognition: Focus on the prefrontal cortex. Pharmacol. Ther. 2007, 113, 523–536.
- Murphy, B.L.; Arnsten, A.F.; Goldman-Rakic, P.S.; Roth, R.H. Increased dopamine turnover in the prefrontal cortex impairs spatial working memory performance in rats and monkeys. Proc. Natl. Acad. Sci. USA 1996, 93, 1325–1329.
- Mizoguchi, K.; Yuzurihara, M.; Ishige, A.; Sasaki, H.; Chui, D.-H.; Tabira, T. Chronic Stress Induces Impairment of Spatial Working Memory Because of Prefrontal Dopaminergic Dysfunction. J. Neurosci. 2000, 20, 1568–1574.
- Lupien, S.J.; Maheu, F.; Tu, M.; Fiocco, A.; Schramek, T.E. The effects of stress and stress hormones on human cognition: Implications for the field of brain and cognition. Brain Cogn. 2007, 65, 209–237.
- Chaouloff, F.; Groc, L. Temporal modulation of hippocampal excitatory transmission by corticosteroids and stress. Front. Neuroendocrinol. 2011, 32, 25–42.
- Gugustea, R.; Jia, Z. Genetic manipulations of AMPA glutamate receptors in hippocampal synaptic plasticity. Neuropharmacology 2021, 194, 108630.
- Kauer, J.A.; Malenka, R.C.; Nicoll, R.A. NMDA application potentiates synaptic transmission in the hippocampus. Nature 1988, 334, 250–252.
- Ortiz, J.B.; Conrad, C.D. The impact from the aftermath of chronic stress on hippocampal structure and function: Is there a recovery? Front. Neuroendocrinol. 2018, 49, 114–123.
- Gould, E.; Tanapat, P. Stress and hippocampal neurogenesis. Biol. Psychiatry 1999, 46, 1472–1479.
- Fuchs, E.; Flügge, G.; Ohl, F.; Lucassen, P.; Vollmann-Honsdorf, G.K.; Michaelis, T. Psychosocial stress, glucocorticoids, and structural alterations in the tree shrew hippocampus. Physiol. Behav. 2001, 73, 285–291.
- Vyas, A.; Pillai, A.G.; Chattarji, S. Recovery after chronic stress fails to reverse amygdaloid neuronal hypertrophy and enhanced anxiety-like behavior. Neuroscience 2004, 128, 667–673.
- Conrad, C.D.; Ortiz, J.B.; Judd, J.M. Chronic stress and hippocampal dendritic complexity: Methodological and functional considerations. Physiol. Behav. 2017, 178, 66–81.
- Zhang, X.; Ge, T.T.; Yin, G.; Cui, R.; Zhao, G.; Yang, W. Stress-Induced Functional Alterations in Amygdala: Implications for Neuropsychiatric Diseases. Front. Neurosci. 2018, 12, 367.
- Winklewski, P.J.; Radkowski, M.; Wszedybyl-Winklewska, M.; Demkow, U. Stress Response, Brain Noradrenergic System and Cognition. Adv. Exp. Med. Biol. 2017, 980, 67–74.
- Liu, W.Z.; Zhang, W.H.; Zheng, Z.H.; Zou, J.X.; Liu, X.X.; Huang, S.H.; You, W.J.; He, Y.; Zhang, J.Y.; Wang, X.D.; et al. Identification of a prefrontal cortex-to-amygdala pathway for chronic stress-induced anxiety. Nat. Commun. 2020, 11, 2221.
- Vyas, A.; Mitra, R.; Shankaranarayana Rao, B.S.; Chattarji, S. Chronic Stress Induces Contrasting Patterns of Dendritic Remodeling in Hippocampal and Amygdaloid Neurons. J. Neurosci. 2002, 22, 6810–6818.
- Sawamura, T.; Klengel, T.; Armario, A.; Jovanovic, T.; Norrholm, S.D.; Ressler, K.J.; Andero, R. Dexamethasone Treatment Leads to Enhanced Fear Extinction and Dynamic Fkbp5 Regulation in Amygdala. Neuropsychopharmacology 2016, 41, 832–846.
- Rainnie, D.G.; Bergeron, R.; Sajdyk, T.J.; Patil, M.; Gehlert, D.R.; Shekhar, A. Corticotrophin releasing factor-induced synaptic plasticity in the amygdala translates stress into emotional disorders. J. Neurosci. 2004, 24, 3471–3479.
- Lankford, K.L.; Letourneau, P.C. Evidence that calcium may control neurite outgrowth by regulating the stability of actin filaments. J. Cell Biol. 1989, 109, 1229–1243.
More
Information
Subjects:
Allergy
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
3.4K
Entry Collection:
Neurodegeneration
Revisions:
2 times
(View History)
Update Date:
07 Sep 2022
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No