 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Kay-Dietrich Wagner | -- | 3585 | 2022-09-03 12:33:53 | | | |
| 2 | Amina Yu | Meta information modification | 3585 | 2022-09-05 03:34:17 | | |
Video Upload Options
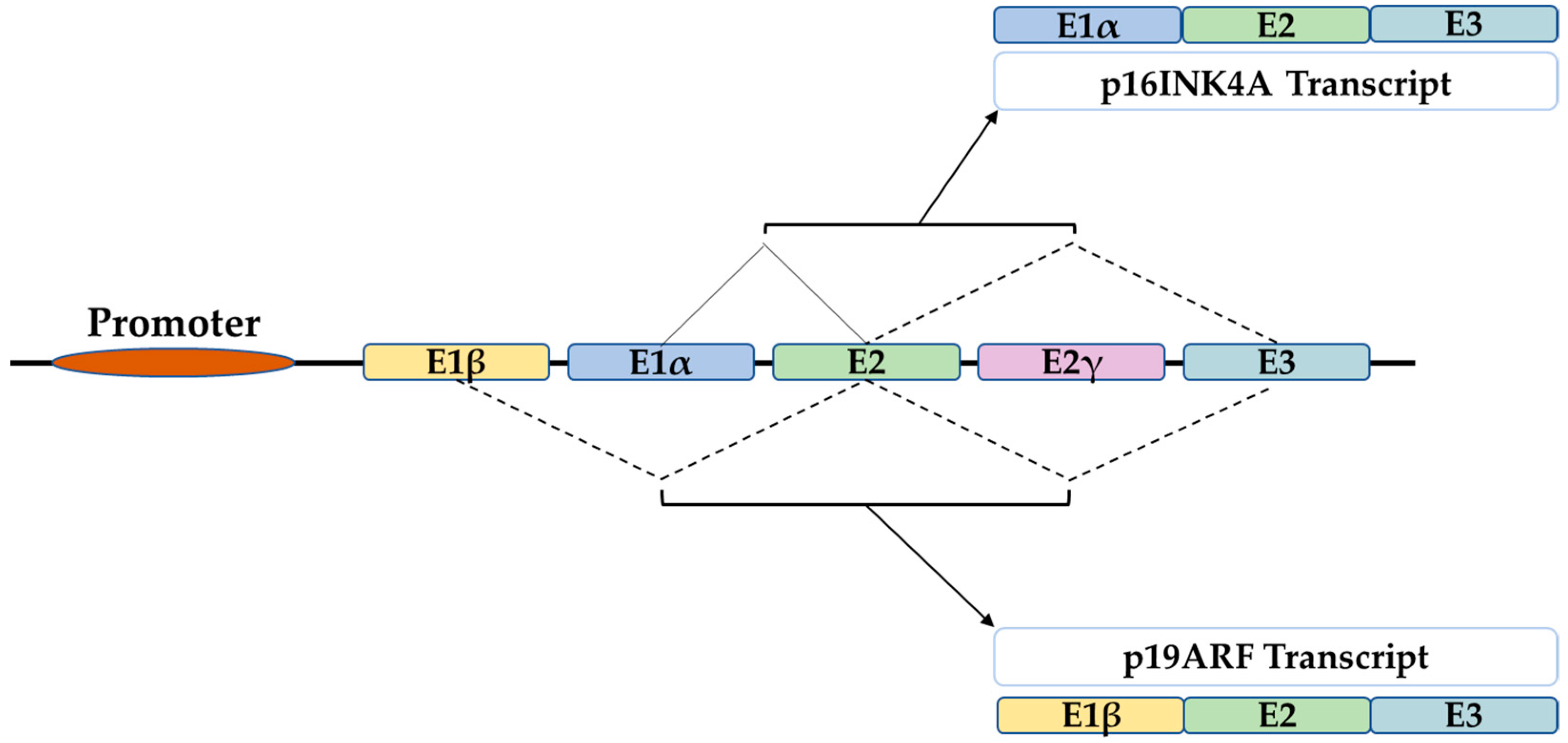
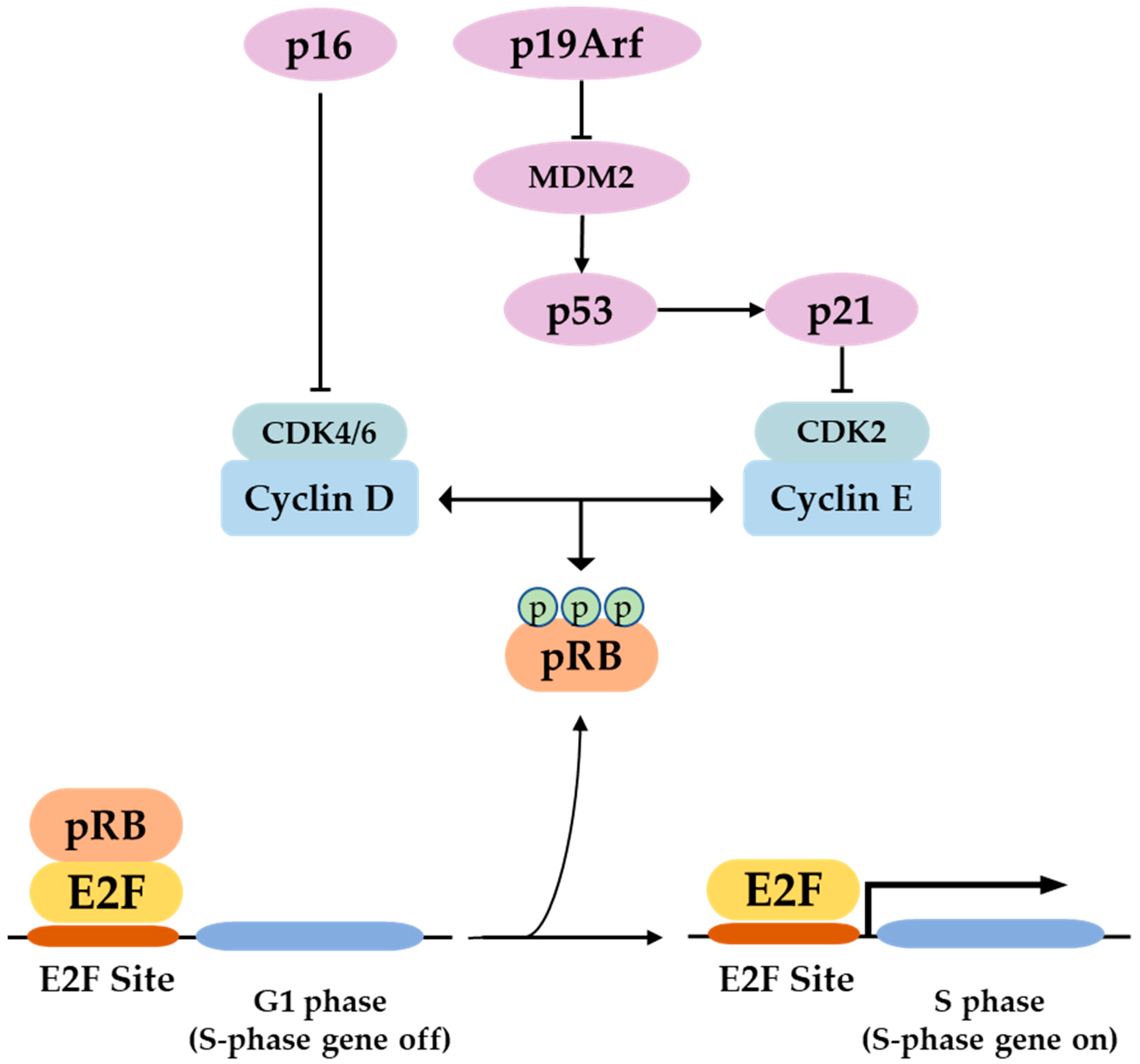
P16 is a tumor suppressor gene, which has been termed with several names such as the multiple tumor suppressor-1 (MTS-1), the inhibitor of cyclin-dependent kinase 4a (INK4A), or the cyclin-dependent kinase inhibitor 2a (CDKN2A). The human p16 gene is located on the short arm of chromosome (9p21.3). The p16 transcript is composed of three exons which encode 156 amino acids. The use of an alternative reading frame generates the human p14Arf protein (p19Arf in mice). In several selected tissues and organs, including skin, bones, lungs, brain, heart, kidney, and liver, it is intended to address the well-known function of p16 in senescence and aging, and discuss several functions of p16, which might be more related to its classical role as a cell cycle regulator.
1. The P16 Gene
2. In the Skin
The skin is the largest tissue in the human body. It serves as a physical barrier to both biological and nonbiological threats. Being exposed to the outside environment places the skin in direct contact with environmental hazards, making it extremely vulnerable. The skin is made up of two layers: the outer epidermis, which is divided into four sublayers with keratinocytes predominating in the spinous, granular, and cornfield sublayers, and pigment-producing melanocytes that confer photoprotection in the basal sublayer. The underlying dermis contains connective tissue with fibroblasts, collagen, and elastin as well as sebaceous and sweat glands and is connected to the epidermis by the dermal epidermal joint (DEJ) [5]. Skin aging is caused by both intrinsic (genetic, time, etc.) and extrinsic (pollution, UV exposure, sunlight, etc.) factors, and it has both biological and functional implications. Aged skin has thinner epidermis, dermis, and DEJ than younger skin, which is due to keratinocytes’ decreased proliferation and renewal ability [6][7][8][9].
3. In the Bones
4. In the Lungs
5. In the Brain
6. In the Heart
7. In the Kidney
8. In the Liver
References
- Serrano, M. The tumor suppressor protein p16INK4a. Exp. Cell Res. 1997, 237, 7–13.
- Komata, T.; Kanzawa, T.; Takeuchi, H.; Germano, I.M.; Schreiber, M.; Kondo, Y.; Kondo, S. Antitumour effect of cyclin-dependent kinase inhibitors (p16(INK4A), p18(INK4C), p19(INK4D), p21(WAF1/CIP1) and p27(KIP1)) on malignant glioma cells. Br. J. Cancer 2003, 88, 1277–1280.
- Li, J.; Poi, M.J.; Tsai, M.-D. Regulatory mechanisms of tumor suppressor P16(INK4A) and their relevance to cancer. Biochemistry 2011, 50, 5566–5582.
- Cilluffo, D.; Barra, V.; Di Leonardo, A. P14ARF: The Absence that Makes the Difference. Genes 2020, 11, 824.
- Blanpain, C.; Fuchs, E. Epidermal Stem Cells of the Skin. Annu. Rev. Cell Dev. Biol. 2006, 22, 339–373.
- Dimri, G.P.; Lee, X.; Basile, G.; Acosta, M.; Scott, G.; Roskelley, C.; Medrano, E.E.; Linskens, M.; Rubelj, I.; Pereira-Smith, O. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. USA 1995, 92, 9363–9367.
- Kligman, A.M. Perspectives and Problems in Cutaneous Gerontology. J. Investig. Dermatol. 1979, 73, 39–46.
- Montagna, W.; Carlisle, K. Structural Changes in Aging Human Skin. J. Investig. Dermatol. 1979, 73, 47–53.
- Mimeault, M.; Batra, S.K. Recent advances on skin-resident stem/progenitor cell functions in skin regeneration, aging and cancers and novel anti-aging and cancer therapies. J. Cell. Mol. Med. 2010, 14, 116–134.
- Debacq-Chainiaux, F.; Borlon, C.; Pascal, T.; Royer, V.; Eliaers, F.; Ninane, N.; Carrard, G.; Friguet, B.; de Longueville, F.; Boffe, S.; et al. Repeated exposure of human skin fibroblasts to UVB at subcytotoxic level triggers premature senescence through the TGF-β1 signaling pathway. J. Cell Sci. 2005, 118, 743–758.
- Lewis, D.A.; Yi, Q.; Travers, J.B.; Spandau, D.F. UVB-induced Senescence in Human Keratinocytes Requires a Functional Insulin-like Growth Factor-1 Receptor and p53. Mol. Biol. Cell 2008, 19, 1346–1353.
- McCart, E.A.; Thangapazham, R.L.; Lombardini, E.D.; Mog, S.R.; Panganiban, R.A.M.; Dickson, K.M.; Mansur, R.A.; Nagy, V.; Kim, S.-Y.; Selwyn, R.; et al. Accelerated senescence in skin in a murine model of radiation-induced multi-organ injury. J. Radiat. Res. 2017, 58, 636–646.
- Nobori, T.; Miura, K.; Wu, D.J.; Lois, A.; Takabayashi, K.; Carson, D.A. Deletions of the cyclin-dependent kinase-4 inhibitor gene in multiple human cancers. Nature 1994, 368, 753–756.
- Scholes, A.G.; Liloglou, T.; Maloney, P.; Hagan, S.; Nunn, J.; Hiscott, P.; Damato, B.E.; Grierson, I.; Field, J.K. Loss of heterozygosity on chromosomes 3, 9, 13, and 17, including the retinoblastoma locus, in uveal melanoma. Investig. Ophthalmol. Vis. Sci. 2001, 42, 2472–2477.
- Castellano, M.; Pollock, P.M.; Walters, M.K.; Sparrow, L.E.; Down, L.M.; Gabrielli, B.G.; Parsons, P.G.; Hayward, N.K. CDKN2A/p16 is inactivated in most melanoma cell lines. Cancer Res. 1997, 57, 4868–4875.
- Funk, J.O.; Schiller, P.I.; Barrett, M.T.; Wong, D.J.; Kind, P.; Sander, C.A. p16INK4a expression is frequently decreased and associated with 9p21 loss of heterozygosity in sporadic melanoma. J. Cutan. Pathol. 1998, 25, 291–296.
- Straume, O.; Sviland, L.; Akslen, L.A. Loss of nuclear p16 protein expression correlates with increased tumor cell proliferation (Ki-67) and poor prognosis in patients with vertical growth phase melanoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2000, 6, 1845–1853.
- Mihic-Probst, D.; Mnich, C.D.; Oberholzer, P.A.; Seifert, B.; Sasse, B.; Moch, H.; Dummer, R. p16 expression in primary malignant melanoma is associated with prognosis and lymph node status. Int. J. Cancer 2006, 118, 2262–2268.
- Tyagi, E.; Liu, B.; Li, C.; Liu, T.; Rutter, J.; Grossman, D. Loss of p16INK4A stimulates aberrant mitochondrial biogenesis through a CDK4/Rb-independent pathway. Oncotarget 2017, 8, 55848–55862.
- Jenkins, N.C.; Liu, T.; Cassidy, P.; Leachman, S.A.; Boucher, K.M.; Goodson, A.G.; Samadashwily, G.; Grossman, D. The p16INK4A tumor suppressor regulates cellular oxidative stress. Oncogene 2011, 30, 265–274.
- Al-Khalaf, H.H.; Mohideen, P.; Nallar, S.C.; Kalvakolanu, D.V.; Aboussekhra, A. The cyclin-dependent kinase inhibitor p16INK4a physically interacts with transcription factor Sp1 and cyclin-dependent kinase 4 to transactivate microRNA-141 and microRNA-146b-5p spontaneously and in response to ultraviolet light-induced DNA damage. J. Biol. Chem. 2013, 288, 35511–35525.
- Demaria, M.; Ohtani, N.; Youssef, S.A.; Rodier, F.; Toussaint, W.; Mitchell, J.R.; Laberge, R.-M.; Vijg, J.; Van Steeg, H.; Dollé, M.E.T.; et al. An Essential Role for Senescent Cells in Optimal Wound Healing through Secretion of PDGF-AA. Dev. Cell 2014, 31, 722–733.
- Jun, J.-I.; Lau, L.F. The matricellular protein CCN1 induces fibroblast senescence and restricts fibrosis in cutaneous wound healing. Nat. Cell Biol. 2010, 12, 676–685.
- Natarajan, E.; Omobono, J.D.; Jones, J.C.; Rheinwald, J.G. Co-expression of p16INK4A and laminin 5 by keratinocytes: A wound-healing response coupling hypermotility with growth arrest that goes awry during epithelial neoplastic progression. J. Investig. Dermatol. Symp. Proc. 2005, 10, 72–85.
- Pavey, S.; Conroy, S.; Russell, T.; Gabrielli, B. Ultraviolet Radiation Induces p16CDKN2A Expression in Human Skin1. Cancer Res. 1999, 59, 4185–4189.
- Adam, R.C.; Fuchs, E. The Yin and Yang of Chromatin Dynamics In Stem Cell Fate Selection. Trends Genet. 2016, 32, 89–100.
- Perdigoto, C.N.; Valdes, V.J.; Bardot, E.S.; Ezhkova, E. Epigenetic regulation of epidermal differentiation. Cold Spring Harb. Perspect. Med. 2014, 4, a015263.
- Botchkarev, V.A.; Gdula, M.R.; Mardaryev, A.N.; Sharov, A.A.; Fessing, M.Y. Epigenetic regulation of gene expression in keratinocytes. J. Investig. Dermatol. 2012, 132, 2505–2521.
- Eckert, R.L.; Adhikary, G.; Rorke, E.A.; Chew, Y.C.; Balasubramanian, S. Polycomb group proteins are key regulators of keratinocyte function. J. Investig. Dermatol. 2011, 131, 295–301.
- Avgustinova, A.; Benitah, S.A. Epigenetic control of adult stem cell function. Nat. Rev. Mol. Cell Biol. 2016, 17, 643–658.
- Safwan-Zaiter, H.; Wagner, N.; Michiels, J.-F.; Wagner, K.-D. Dynamic Spatiotemporal Expression Pattern of the Senescence-Associated Factor p16Ink4a in Development and Aging. Cells 2022, 11, 541.
- Langlands, K.; Down, G.A.; Kealey, T. Id proteins are dynamically expressed in normal epidermis and dysregulated in squamous cell carcinoma. Cancer Res. 2000, 60, 5929–5933.
- D’Arcangelo, D.; Tinaburri, L.; Dellambra, E. The Role of p16INK4a Pathway in Human Epidermal Stem Cell Self-Renewal, Aging and Cancer. Int. J. Mol. Sci. 2017, 18, 1591.
- Chen, X.; Wang, Z.; Duan, N.; Zhu, G.; Schwarz, E.M.; Xie, C. Osteoblast-Osteoclast Interactions. Connect. Tissue Res. 2018, 59, 99–107.
- Jilka, R.L.; O’Brien, C.A. The Role of Osteocytes in Age-Related Bone Loss. Curr. Osteoporos. Rep. 2016, 14, 16–25.
- Almeida, M.; Han, L.; Martin-Millan, M.; Plotkin, L.I.; Stewart, S.A.; Roberson, P.K.; Kousteni, S.; O’Brien, C.A.; Bellido, T.; Parfitt, A.M.; et al. Skeletal involution by age-associated oxidative stress and its acceleration by loss of sex steroids. J. Biol. Chem. 2007, 282, 27285–27297.
- Glatt, V.; Canalis, E.; Stadmeyer, L.; Bouxsein, M.L. Age-related changes in trabecular architecture differ in female and male C57BL/6J mice. J. Bone Miner. Res. 2007, 22, 1197–1207.
- Piemontese, M.; Almeida, M.; Robling, A.G.; Kim, H.-N.; Xiong, J.; Thostenson, J.D.; Weinstein, R.S.; Manolagas, S.C.; O’Brien, C.A.; Jilka, R.L. Old age causes de novo intracortical bone remodeling and porosity in mice. JCI Insight 2017, 2, 93771.
- Ucer, S.; Iyer, S.; Kim, H.-N.; Han, L.; Rutlen, C.; Allison, K.; Thostenson, J.D.; de Cabo, R.; Jilka, R.L.; O’Brien, C.; et al. The Effects of Aging and Sex Steroid Deficiency on the Murine Skeleton Are Independent and Mechanistically Distinct. J. Bone Miner. Res. 2017, 32, 560–574.
- Marie, P.J. Bone Cell Senescence: Mechanisms and Perspectives. J. Bone Miner. Res. 2014, 29, 1311–1321.
- Baron, R.; Kneissel, M. WNT signaling in bone homeostasis and disease: From human mutations to treatments. Nat. Med. 2013, 19, 179–192.
- Farr, J.N.; Fraser, D.G.; Wang, H.; Jaehn, K.; Ogrodnik, M.B.; Weivoda, M.M.; Drake, M.T.; Tchkonia, T.; LeBrasseur, N.K.; Kirkland, J.L.; et al. Identification of Senescent Cells in the Bone Microenvironment. J. Bone Miner. Res. 2016, 31, 1920–1929.
- Kim, H.-N.; Chang, J.; Shao, L.; Han, L.; Iyer, S.; Manolagas, S.C.; O’Brien, C.A.; Jilka, R.L.; Zhou, D.; Almeida, M. DNA damage and senescence in osteoprogenitors expressing Osx1 may cause their decrease with age. Aging Cell 2017, 16, 693–703.
- Farr, J.N.; Xu, M.; Weivoda, M.M.; Monroe, D.G.; Fraser, D.G.; Onken, J.L.; Negley, B.A.; Sfeir, J.G.; Ogrodnik, M.B.; Hachfeld, C.M.; et al. Targeting cellular senescence prevents age-related bone loss in mice. Nat. Med. 2017, 23, 1072–1079.
- Kim, H.-N.; Chang, J.; Iyer, S.; Han, L.; Campisi, J.; Manolagas, S.C.; Zhou, D.; Almeida, M. Elimination of senescent osteoclast progenitors has no effect on the age-associated loss of bone mass in mice. Aging Cell 2019, 18, e12923.
- Peng, S.; Chen, X.; Huang, C.; Yang, C.; Situ, M.; Zhou, Q.; Ling, Y.; Huang, H.; Huang, M.; Zhang, Y.; et al. UBE2S as a novel ubiquitinated regulator of p16 and β-catenin to promote bone metastasis of prostate cancer. Int. J. Biol. Sci. 2022, 18, 3528–3543.
- Harris, A.S.; Thomas, R.G.; Passant, C.D. Do patients with p16-positive oropharyngeal squamous cell carcinoma get more bone metastasis than p16-negative patients? J. Laryngol. Otol. 2018, 132, 429–433.
- Righi, A.; Gambarotti, M.; Sbaraglia, M.; Sisto, A.; Ferrari, S.; Dei Tos, A.P.; Picci, P. p16 expression as a prognostic and predictive marker in high-grade localized osteosarcoma of the extremities: An analysis of 357 cases. Hum. Pathol. 2016, 58, 15–23.
- Chandra, A.; Lagnado, A.B.; Farr, J.N.; Doolittle, M.; Tchkonia, T.; Kirkland, J.L.; LeBrasseur, N.K.; Robbins, P.D.; Niedernhofer, L.J.; Ikeno, Y.; et al. Targeted clearance of p21- but not p16-positive senescent cells prevents radiation-induced osteoporosis and increased marrow adiposity. Aging Cell 2022, 21, e13602.
- Li, J.; Karim, M.A.; Che, H.; Geng, Q.; Miao, D. Deletion of p16 prevents estrogen deficiency-induced osteoporosis by inhibiting oxidative stress and osteocyte senescence. Am. J. Transl. Res. 2020, 12, 672–683.
- Ding, Q.; Liu, H.; Liu, L.; Ma, C.; Qin, H.; Wei, Y.; Ren, Y. Deletion of p16 accelerates fracture healing in geriatric mice. Am. J. Transl. Res. 2021, 13, 11107.
- Mercado, N.; Ito, K.; Barnes, P.J. Accelerated ageing of the lung in COPD: New concepts. Thorax 2015, 70, 482–489.
- Fukuchi, Y. The aging lung and chronic obstructive pulmonary disease: Similarity and difference. Proc. Am. Thorac. Soc. 2009, 6, 570–572.
- John-Schuster, G.; Günter, S.; Hager, K.; Conlon, T.M.; Eickelberg, O.; Yildirim, A.Ö. Inflammaging increases susceptibility to cigarette smoke-induced COPD. Oncotarget 2016, 7, 30068–30083.
- Meiners, S.; Eickelberg, O.; Königshoff, M. Hallmarks of the ageing lung. Eur. Respir. J. 2015, 45, 807–827.
- Selman, M.; Buendía-Roldán, I.; Pardo, A. Aging and Pulmonary Fibrosis. Rev. Investig. Clin. 2016, 68, 75–83.
- Venosa, A. Senescence in Pulmonary Fibrosis: Between Aging and Exposure. Front. Med. 2020, 7, 606462.
- Kheradmand, F.; You, R.; Hee Gu, B.; Corry, D.B. Cigarette Smoke and DNA Cleavage Promote Lung Inflammation and Emphysema. Trans. Am. Clin. Climatol. Assoc. 2017, 128, 222–233.
- Nyunoya, T.; Monick, M.M.; Klingelhutz, A.L.; Glaser, H.; Cagley, J.R.; Brown, C.O.; Matsumoto, E.; Aykin-Burns, N.; Spitz, D.R.; Oshima, J.; et al. Cigarette Smoke Induces Cellular Senescence via Werner’s Syndrome Protein Down-regulation. Am. J. Respir. Crit. Care Med. 2009, 179, 279–287.
- Nyunoya, T.; Monick, M.M.; Klingelhutz, A.; Yarovinsky, T.O.; Cagley, J.R.; Hunninghake, G.W. Cigarette Smoke Induces Cellular Senescence. Am. J. Respir. Cell Mol. Biol. 2006, 35, 681–688.
- Rashid, K.; Sundar, I.K.; Gerloff, J.; Li, D.; Rahman, I. Lung cellular senescence is independent of aging in a mouse model of COPD/emphysema. Sci. Rep. 2018, 8, 9023.
- Cottage, C.T.; Peterson, N.; Kearley, J.; Berlin, A.; Xiong, X.; Huntley, A.; Zhao, W.; Brown, C.; Migneault, A.; Zerrouki, K.; et al. Targeting p16-induced senescence prevents cigarette smoke-induced emphysema by promoting IGF1/Akt1 signaling in mice. Commun. Biol. 2019, 2, 307.
- Wang, C.-W.; Wu, T.-I.; Yu, C.-T.; Wu, Y.-C.; Teng, Y.-H.; Chin, S.-Y.; Lai, C.-H.; Chen, T.-C. Usefulness of p16 for differentiating primary pulmonary squamous cell carcinoma from cervical squamous cell carcinoma metastatic to the lung. Am. J. Clin. Pathol. 2009, 131, 715–722.
- Belinsky, S.A.; Nikula, K.J.; Palmisano, W.A.; Michels, R.; Saccomanno, G.; Gabrielson, E.; Baylin, S.B.; Herman, J.G. Aberrant methylation of p16(INK4a) is an early event in lung cancer and a potential biomarker for early diagnosis. Proc. Natl. Acad. Sci. USA 1998, 95, 11891–11896.
- Okamoto, A.; Hussain, S.P.; Hagiwara, K.; Spillare, E.A.; Rusin, M.R.; Demetrick, D.J.; Serrano, M.; Hannon, G.J.; Shiseki, M.; Zariwala, M.; et al. Mutations in the p16INK4/MTS1/CDKN2, p15INK4B/MTS2, and p18 Genes in Primary and Metastatic Lung Cancer1. Cancer Res. 1995, 55, 1448–1451.
- Tong, J.; Sun, X.; Cheng, H.; Zhao, D.; Ma, J.; Zhen, Q.; Cao, Y.; Zhu, H.; Bai, J. Expression of p16 in non-small cell lung cancer and its prognostic significance: A meta-analysis of published literatures. Lung Cancer 2011, 74, 155–163.
- p16 Regulation of Lung Epithelial Cell Growth, Repair after Injury and Transformation—ProQuest. Available online: https://www.proquest.com/openview/c4b38b5ea3ce9a758bbacff031039f5c/1?pq-origsite=gscholar&cbl=18750&diss=y (accessed on 28 July 2022).
- De Mochel, N.R.; Cheong, K.N.; Cassandras, M.; Wang, C.; Krasilnikov, M.; Matatia, P.; Molofsky, A.; Campisi, J.; Peng, T. Sentinel p16INK4a+ cells in the basement membrane form a reparative niche in the lung 2020. bioRxiv 2020.
- Molofsky, A.V.; Slutsky, S.G.; Joseph, N.M.; He, S.; Pardal, R.; Krishnamurthy, J.; Sharpless, N.E.; Morrison, S.J. Increasing p16INK4a expression decreases forebrain progenitors and neurogenesis during ageing. Nature 2006, 443, 448–452.
- Song, P.; An, J.; Zou, M.-H. Immune Clearance of Senescent Cells to Combat Ageing and Chronic Diseases. Cells 2020, 9, 671.
- Lok, K.; Zhao, H.; Shen, H.; Wang, Z.; Gao, X.; Zhao, W.; Yin, M. Characterization of the APP/PS1 mouse model of Alzheimer’s disease in senescence accelerated background. Neurosci. Lett. 2013, 557, 84–89.
- Dorigatti, A.O.; Riordan, R.; Yu, Z.; Ross, G.; Wang, R.; Reynolds-Lallement, N.; Magnusson, K.; Galvan, V.; Perez, V.I. Brain cellular senescence in mouse models of Alzheimer’s disease. GeroScience 2022, 44, 1157–1168.
- Musi, N.; Valentine, J.M.; Sickora, K.R.; Baeuerle, E.; Thompson, C.S.; Shen, Q.; Orr, M.E. Tau protein aggregation is associated with cellular senescence in the brain. Aging Cell 2018, 17, e12840.
- Ramsden, M.; Kotilinek, L.; Forster, C.; Paulson, J.; McGowan, E.; SantaCruz, K.; Guimaraes, A.; Yue, M.; Lewis, J.; Carlson, G.; et al. Age-Dependent Neurofibrillary Tangle Formation, Neuron Loss, and Memory Impairment in a Mouse Model of Human Tauopathy (P301L). J. Neurosci. 2005, 25, 10637–10647.
- Mv, S.; Hv, V. Astrocytes: Biology and pathology. Acta Neuropathol. 2010, 119, 7–35.
- Halassa, M.M.; Fellin, T.; Haydon, P.G. Tripartite synapses: Roles for astrocytic purines in the control of synaptic physiology and behavior. Neuropharmacology 2009, 57, 343–346.
- Pertusa, M.; García-Matas, S.; Rodriguez-Farre, E.; Sanfeliu, C.; Cristòfol, R. Astrocytes aged in vitro show a decreased neuroprotective capacity. J. Neurochem. 2007, 101, 794–805.
- Cohen, J.; Torres, C. Astrocyte senescence: Evidence and significance. Aging Cell 2019, 18, e12937.
- Jen, J.; Harper, J.W.; Bigner, S.H.; Bigner, D.D.; Papadopoulos, N.; Markowitz, S.; Willson, J.K.V.; Kinzler, K.W.; Vogelstein, B. Deletion of p16 and p15 Genes in Brain Tumors1. Cancer Res. 1994, 54, 6353–6358.
- Ueki, K.; Ono, Y.; Henson, J.W.; Efird, J.T.; von Deimling, A.; Louis, D.N. CDKN2/p16 or RB Alterations Occur in the Majority of Glioblastomas and Are Inversely Correlated1. Cancer Res. 1996, 56, 150–153.
- Park, J.W.; Kang, J.; Lim, K.Y.; Kim, H.; Kim, S.-I.; Won, J.K.; Park, C.-K.; Park, S.-H. The prognostic significance of p16 expression pattern in diffuse gliomas. J. Pathol. Transl. Med. 2021, 55, 102–111.
- Fueyo, J.; Gomez-Manzano, C.; Puduvalli, V.K.; Martin-Duque, P.; Perez-Soler, R.; Levin, V.A.; Yung, W.K.; Kyritsis, A.P. Adenovirus-mediated p16 transfer to glioma cells induces G1 arrest and protects from paclitaxel and topotecan: Implications for therapy. Int. J. Oncol. 1998, 12, 665–674.
- Kranenburg, O.; van der Eb, A.J.; Zantema, A. Cyclin D1 is an essential mediator of apoptotic neuronal cell death. EMBO J. 1996, 15, 46–54.
- Kfoury, N.; Sun, T.; Yu, K.; Rockwell, N.; Tinkum, K.L.; Qi, Z.; Warrington, N.M.; McDonald, P.; Roy, A.; Weir, S.J.; et al. Cooperative p16 and p21 action protects female astrocytes from transformation. Acta Neuropathol. Commun. 2018, 6, 12.
- Yabluchanskiy, A.; Tarantini, S.; Balasubramanian, P.; Kiss, T.; Csipo, T.; Fülöp, G.A.; Lipecz, A.; Ahire, C.; DelFavero, J.; Nyul-Toth, A.; et al. Pharmacological or genetic depletion of senescent astrocytes prevents whole brain irradiation–induced impairment of neurovascular coupling responses protecting cognitive function in mice. GeroScience 2020, 42, 409–428.
- Qian, J.; Wang, X.; Cao, J.; Zhang, W.; Lu, C.; Chen, X. Dihydromyricetin attenuates D-galactose-induced brain aging of mice via inhibiting oxidative stress and neuroinflammation. Neurosci. Lett. 2021, 756, 135963.
- Torella, D.; Rota, M.; Nurzynska, D.; Musso, E.; Monsen, A.; Shiraishi, I.; Zias, E.; Walsh, K.; Rosenzweig, A.; Sussman, M.A.; et al. Cardiac stem cell and myocyte aging, heart failure, and insulin-like growth factor-1 overexpression. Circ. Res. 2004, 94, 514–524.
- Chimenti, C.; Kajstura, J.; Torella, D.; Urbanek, K.; Heleniak, H.; Colussi, C.; Di Meglio, F.; Nadal-Ginard, B.; Frustaci, A.; Leri, A.; et al. Senescence and Death of Primitive Cells and Myocytes Lead to Premature Cardiac Aging and Heart Failure. Circ. Res. 2003, 93, 604–613.
- Anderson, R.; Lagnado, A.; Maggiorani, D.; Walaszczyk, A.; Dookun, E.; Chapman, J.; Birch, J.; Salmonowicz, H.; Ogrodnik, M.; Jurk, D.; et al. Length-independent telomere damage drives post-mitotic cardiomyocyte senescence. EMBO J. 2019, 38, e100492.
- Lewis-McDougall, F.C.; Ruchaya, P.J.; Domenjo-Vila, E.; Shin Teoh, T.; Prata, L.; Cottle, B.J.; Clark, J.E.; Punjabi, P.P.; Awad, W.; Torella, D.; et al. Aged-senescent cells contribute to impaired heart regeneration. Aging Cell 2019, 18, e12931.
- Kajstura, J.; Pertoldi, B.; Leri, A.; Beltrami, C.-A.; Deptala, A.; Darzynkiewicz, Z.; Anversa, P. Telomere Shortening Is an in Vivo Marker of Myocyte Replication and Aging. Am. J. Pathol. 2000, 156, 813–819.
- Matthews, C.; Gorenne, I.; Scott, S.; Figg, N.; Kirkpatrick, P.; Ritchie, A.; Goddard, M.; Bennett, M. Vascular Smooth Muscle Cells Undergo Telomere-Based Senescence in Human Atherosclerosis. Circ. Res. 2006, 99, 156–164.
- Cianflone, E.; Torella, M.; Biamonte, F.; De Angelis, A.; Urbanek, K.; Costanzo, F.S.; Rota, M.; Ellison-Hughes, G.M.; Torella, D. Targeting Cardiac Stem Cell Senescence to Treat Cardiac Aging and Disease. Cells 2020, 9, 1558.
- Epstein, J.A. A Time to Press Reset and Regenerate Cardiac Stem Cell Biology. JAMA Cardiol. 2019, 4, 95–96.
- Baker, D.J.; Childs, B.G.; Durik, M.; Wijers, M.E.; Sieben, C.J.; Zhong, J.; Saltness, R.A.; Jeganathan, K.B.; Verzosa, G.C.; Pezeshki, A.; et al. Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature 2016, 530, 184–189.
- Grosse, L.; Wagner, N.; Emelyanov, A.; Molina, C.; Lacas-Gervais, S.; Wagner, K.-D.; Bulavin, D.V. Defined p16High Senescent Cell Types Are Indispensable for Mouse Healthspan. Cell Metab. 2020, 32, 87–99e6.
- Shi, J.; Sun, J.; Liu, L.; Shan, T.; Meng, H.; Yang, T.; Wang, S.; Wei, T.; Chen, B.; Ma, Y.; et al. P16ink4a overexpression ameliorates cardiac remodeling of mouse following myocardial infarction via CDK4/pRb pathway. Biochem. Biophys. Res. Commun. 2022, 595, 62–68.
- Joosten, S.A.; van Ham, V.; Nolan, C.E.; Borrias, M.C.; Jardine, A.G.; Shiels, P.G.; van Kooten, C.; Paul, L.C. Telomere Shortening and Cellular Senescence in a Model of Chronic Renal Allograft Rejection. Am. J. Pathol. 2003, 162, 1305–1312.
- Chkhotua, A.B.; Gabusi, E.; Altimari, A.; D’Errico, A.; Yakubovich, M.; Vienken, J.; Stefoni, S.; Chieco, P.; Yussim, A.; Grigioni, W.F. Increased expression of p16(INK4a) and p27(Kip1) cyclin-dependent kinase inhibitor genes in aging human kidney and chronic allograft nephropathy. Am. J. Kidney Dis. 2003, 41, 1303–1313.
- Melk, A.; Schmidt, B.M.W.; Vongwiwatana, A.; Rayner, D.C.; Halloran, P.F. Increased Expression of Senescence-Associated Cell Cycle Inhibitor p16INK4a in Deteriorating Renal Transplants and Diseased Native Kidney. Am. J. Transplant. 2005, 5, 1375–1382.
- Melk, A.; Schmidt, B.M.W.; Takeuchi, O.; Sawitzki, B.; Rayner, D.C.; Halloran, P.F. Expression of p16INK4a and other cell cycle regulator and senescence associated genes in aging human kidney. Kidney Int. 2004, 65, 510–520.
- Sis, B.; Tasanarong, A.; Khoshjou, F.; Dadras, F.; Solez, K.; Halloran, P.F. Accelerated expression of senescence associated cell cycle inhibitor p16INK4A in kidneys with glomerular disease. Kidney Int. 2007, 71, 218–226.
- Westhoff, J.H.; Hilgers, K.F.; Steinbach, M.P.; Hartner, A.; Klanke, B.; Amann, K.; Melk, A. Hypertension induces somatic cellular senescence in rats and humans by induction of cell cycle inhibitor p16INK4a. Hypertension 2008, 52, 123–129.
- Liu, J.; Yang, J.-R.; Chen, X.-M.; Cai, G.-Y.; Lin, L.-R.; He, Y.-N. Impact of ER stress-regulated ATF4/p16 signaling on the premature senescence of renal tubular epithelial cells in diabetic nephropathy. Am. J. Physiol.-Cell Physiol. 2015, 308, C621–C630.
- Gu, X.; Peng, C.-Y.; Lin, S.-Y.; Qin, Z.-Y.; Liang, J.-L.; Chen, H.-J.; Hou, C.-X.; Wang, R.; Du, Y.-Q.; Jin, J.-L.; et al. P16INK4a played a critical role in exacerbating acute tubular necrosis in acute kidney injury. Am. J. Transl. Res. 2019, 11, 3850–3861.
- Jin, J.; Tao, J.; Gu, X.; Yu, Z.; Wang, R.; Zuo, G.; Li, Q.; Lv, X.; Miao, D. P16 INK4a Deletion Ameliorated Renal Tubulointerstitial Injury in a Stress-induced Premature Senescence Model of Bmi-1 Deficiency. Sci. Rep. 2017, 7, 7502.
- Baiocchi, L.; Glaser, S.; Francis, H.; Kennedy, L.; Felli, E.; Alpini, G.; Gracia-Sancho, J. Impact of Aging on Liver Cells and Liver Disease: Focus on the Biliary and Vascular Compartments. Hepatol. Commun. 2021, 5, 1125–1137.
- Kim, H.; Kisseleva, T.; Brenner, D.A. Aging and liver disease. Curr. Opin. Gastroenterol. 2015, 31, 184–191.
- Aravinthan, A.D.; Alexander, G.J.M. Senescence in chronic liver disease: Is the future in aging? J. Hepatol. 2016, 65, 825–834.
- Zhu, C.; Ikemoto, T.; Utsunomiya, T.; Yamada, S.; Morine, Y.; Imura, S.; Arakawa, Y.; Takasu, C.; Ishikawa, D.; Shimada, M. Senescence-related genes possibly responsible for poor liver regeneration after hepatectomy in elderly patients. J. Gastroenterol. Hepatol. 2014, 29, 1102–1108.
- Sawada, N. Hepatocytes from old rats retain responsiveness of c-myc expression to EGF in primary culture but do not enter S phase. Exp. Cell Res. 1989, 181, 584–588.
- Maeso-Díaz, R.; Ortega-Ribera, M.; Fernández-Iglesias, A.; Hide, D.; Muñoz, L.; Hessheimer, A.J.; Vila, S.; Francés, R.; Fondevila, C.; Albillos, A.; et al. Effects of aging on liver microcirculatory function and sinusoidal phenotype. Aging Cell 2018, 17, e12829.
- Omori, S.; Wang, T.-W.; Johmura, Y.; Kanai, T.; Nakano, Y.; Kido, T.; Susaki, E.A.; Nakajima, T.; Shichino, S.; Ueha, S.; et al. Generation of a p16 Reporter Mouse and Its Use to Characterize and Target p16high Cells In Vivo. Cell Metab. 2020, 32, 814–828e6.
- González-Navarro, H.; Vinué, Á.; Sanz, M.J.; Delgado, M.; Pozo, M.A.; Serrano, M.; Burks, D.J.; Andrés, V. Increased dosage of Ink4/Arf protects against glucose intolerance and insulin resistance associated with aging. Aging Cell 2013, 12, 102–111.
- Pal, A.; Potjer, T.P.; Thomsen, S.K.; Ng, H.J.; Barrett, A.; Scharfmann, R.; James, T.J.; Bishop, D.T.; Karpe, F.; Godsland, I.F.; et al. Loss-of-Function Mutations in the Cell-Cycle Control Gene CDKN2A Impact on Glucose Homeostasis in Humans. Diabetes 2016, 65, 527–533.
- Bantubungi, K.; Hannou, S.-A.; Caron-Houde, S.; Vallez, E.; Baron, M.; Lucas, A.; Bouchaert, E.; Paumelle, R.; Tailleux, A.; Staels, B. Cdkn2a/p16Ink4a regulates fasting-induced hepatic gluconeogenesis through the PKA-CREB-PGC1α pathway. Diabetes 2014, 63, 3199–3209.
- Deleye, Y.; Cotte, A.K.; Hannou, S.A.; Hennuyer, N.; Bernard, L.; Derudas, B.; Caron, S.; Legry, V.; Vallez, E.; Dorchies, E.; et al. CDKN2A/p16INK4a suppresses hepatic fatty acid oxidation through the AMPKα2-SIRT1-PPARα signaling pathway. J. Biol. Chem. 2020, 295, 17310–17322.
- Ogrodnik, M.; Miwa, S.; Tchkonia, T.; Tiniakos, D.; Wilson, C.L.; Lahat, A.; Day, C.P.; Burt, A.; Palmer, A.; Anstee, Q.M.; et al. Cellular senescence drives age-dependent hepatic steatosis. Nat. Commun. 2017, 8, 15691.
- Zhang, X.; Xu, G.B.; Zhou, D.; Pan, Y.-X. High-fat diet modifies expression of hepatic cellular senescence gene p16(INK4a) through chromatin modifications in adult male rats. Genes Nutr. 2018, 13, 6.
- Zang, J.-J.; Xie, F.; Xu, J.-F.; Qin, Y.-Y.; Shen, R.-X.; Yang, J.-M.; He, J. P16 gene hypermethylation and hepatocellular carcinoma: A systematic review and meta-analysis. World J. Gastroenterol. 2011, 17, 3043–3048.
- Wong, I.H.N.; Dennis Lo, Y.M.; Zhang, J.; Liew, C.-T.; Ng, M.H.L.; Wong, N.; Lai, P.B.S.; Lau, W.Y.; Hjelm, N.M.; Johnson, P.J. Detection of Aberrant p16 Methylation in the Plasma and Serum of Liver Cancer Patients1. Cancer Res. 1999, 59, 71–73.


