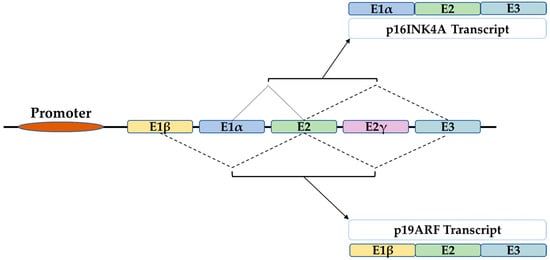
Figure 1. The structure of the INK4A locus gives rise to several transcripts through alternative splicing. P16INK4A and p19ARF are two transcripts of 3 exons which differ only in the first exon that is E1α for p16 and E1β for p19ARF.
2. In the Skin
The skin is the largest tissue in the human body. It serves as a physical barrier to both biological and nonbiological threats. Being exposed to the outside environment places the skin in direct contact with environmental hazards, making it extremely vulnerable. The skin is made up of two layers: the outer epidermis, which is divided into four sublayers with keratinocytes predominating in the spinous, granular, and cornfield sublayers, and pigment-producing melanocytes that confer photoprotection in the basal sublayer. The underlying dermis contains connective tissue with fibroblasts, collagen, and elastin as well as sebaceous and sweat glands and is connected to the epidermis by the dermal epidermal joint (DEJ) [5]. Skin aging is caused by both intrinsic (genetic, time, etc.) and extrinsic (pollution, UV exposure, sunlight, etc.) factors, and it has both biological and functional implications. Aged skin has thinner epidermis, dermis, and DEJ than younger skin, which is due to keratinocytes’ decreased proliferation and renewal ability [6][7][8][9].
2. In the Skin
The skin is the largest tissue in the human body. It serves as a physical barrier to both biological and nonbiological threats. Being exposed to the outside environment places the skin in direct contact with environmental hazards, making it extremely vulnerable. The skin is made up of two layers: the outer epidermis, which is divided into four sublayers with keratinocytes predominating in the spinous, granular, and cornfield sublayers, and pigment-producing melanocytes that confer photoprotection in the basal sublayer. The underlying dermis contains connective tissue with fibroblasts, collagen, and elastin as well as sebaceous and sweat glands and is connected to the epidermis by the dermal epidermal joint (DEJ) [98]. Skin aging is caused by both intrinsic (genetic, time, etc.) and extrinsic (pollution, UV exposure, sunlight, etc.) factors, and it has both biological and functional implications. Aged skin has thinner epidermis, dermis, and DEJ than younger skin, which is due to keratinocytes’ decreased proliferation and renewal ability [25,99,100,101].
As major biomarkers of senescence, both the SA-β-gal and p16 determination has shown elevated expression upon in vitro exposure of fibroblasts and keratinocytes to UV light
[10][11][12][102,103,104]. Furthermore, telomere shortening, DNA damage, and UV exposure increased the activity of the p16/pRB and P19ARF/p53/P21 cascades, resulting in an accumulation of senescent cells and skin stem cell dysfunction and loss of regeneration capacity
[9][101]. An in vivo study, on the other hand, claimed that UV light exposure has accelerated cellular senescence by increasing p21 expression
[12][104].
In contrast to the previous results, the presence of p16 has been shown to play an important role in several biological processes that are beneficial to the skin. Starting with its tumor suppression function, p16 inactivation due to mutation or promoter methylation has been linked to a variety of cancers, including familial and sporadic melanoma
[13][14][15][16][105,106,107,108]. These studies identified 55 out of 60 melanoma cell lines that were dependent on complete or partial p16 aberration, implicating this pathway in the development of melanomas. Furthermore, the level of p16 expression could be used as a melanoma predictive and prognostic biomarker. In other words, lower p16 levels were associated with higher Ki67 expression as a proliferation marker, and metastatic melanoma lesions were associated with even lower p16 levels and predicted poor patient survival
[17][109]. Benign nevi had higher p16 levels than nonmetastatic melanoma, which had even higher p16 levels than metastatic melanoma
[18][110]. Furthermore, in primary mouse fibroblasts (PMFs), human melanocytes, and a human melanoma cell line (A375), the loss of p16 correlated with increased mitochondrial mass, attenuated respiration, and altered morphology associated with augmented superoxide production and higher cellular motility. Forced p16 expression restored mitochondrial homeostasis, dynamics, and motility in a CDK4/pRB independent pathway
[19][111]. Surprisingly, oxidative stress-induced p16 has attenuated ROS production in skin in vivo and in vitro. In addition, elevated intracellular ROS and DNA damage were obtained in p16-deficient cells. This was restored in skin fibroblasts transduced with p16 using lentivirus
[20][72]. These findings suggest a pRB-independent tumor suppression function of p16. As another mechanism, p16 has been found to transactivate the promoter of the tumor suppressor miRNAs, miRNA-141 and miRNA-146b-5p, in melanocyte through physical interaction with the transcription factor Sp1 and CDK4, via the p16 fourth ankyrin repeat. Mutation in this ankyrin repeat attenuated Sp1 binding and miRNA-141 and miRNA-146b-5p transactivation without affecting the expression level of Sp1
[21][112]. In addition, this p16–Sp1–CDK4 interaction and consequent miRNA-141 and miRNA-146b-5p transactivation has also been implicated in cellular response to UV-radiation-induced damage and apoptosis.
P16 has been shown to be an important factor in wound healing. Endothelial cells and fibroblasts were identified as p16-positive cells at the site of injury in the p16-3MR mouse model a few days after injury. These transiently appearing senescent cells aimed to accelerate wound closure by inducing myofibroblast differentiation via platelet-derived growth factor AA secretion as part of the SASP
[22][43]. Elimination of these cells delayed the wound healing process. The matricellular protein CCN1 has been identified as a key player in the induction of fibroblast senescence at the wound healing margins. By inducing DNA damage and p53 activity, CCN1 induces oxidative stress and provokes p16 upregulation, which leads to fibroblast senescence and antifibrotic gene activation
[23][113]. Furthermore, coexpression and activation of the laminin 5/p16 response has been identified in migrating keratinocytes. The laminin 5/p16 response caused hypermotility and growth arrest in keratinocytes, leading to wound re-epithelialization
[24][44]. This pathway has also been identified in critical stage neoplastic progression as a tumor suppressing pathway. This might suggest a protective effect of the induced p16 upregulation upon the exposure of skin to UV radiation
[25][114].
Moreover, p16-orchestrated expression is required for stem cell self-renewal and differentiation. More precisely, p16 repression by epigenetic regulators is indispensable for stem cells proliferation. On the contrary, its promoter epigenetic regulation and orchestrated expression level have been found crucial for keratinocyte differentiation beside many other differentiation genes
[26][27][28][29][30][115,116,117,118,119]. However, p16 seems not causal for terminal differentiation as it is expressed during early embryonic development
[31][120], but still the balance between growth and differentiation requires a balanced expression of p16 and other cell cycle regulators
[27][28][29][30][116,117,118,119]. For instance, Id-1, Id-2, and Id-3 are repressors of p16 and are upregulated in dividing keratinocytes, whereas they become downregulated in differentiated cells
[32][121]. Activators of p16 transcription promoted keratinocyte differentiation via acting on epidermal differentiation complex genes
[33][46]. Therefore, unravelling the precise mechanism underlying p16 regulation of expression might provide a targeted approach which confers maintenance of epidermis regenerative capacity and avoids premature skin aging or cancer development (
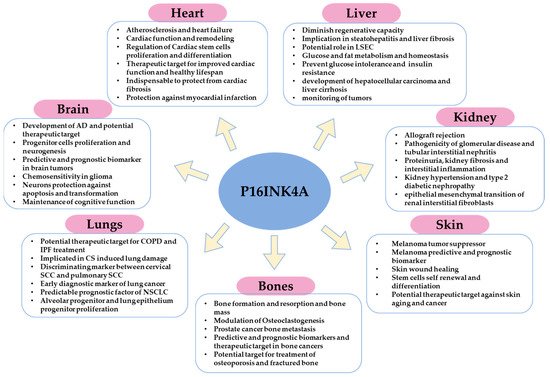
Figure 23).
Figure 23. Schematic illustration that summarizes the major functions or implications of p16 in homeostasis, pathophysiology, and cancer of different organs.
3. In the Bones
Two major types of cells are involved in maintaining skeletal homeostasis: osteoblasts, which are derived from osteoprogenitor cells and are in charge of bone growth, mineralization, and remodeling, and osteoclasts, which are descended from myeloid lineages and mediate bone resorption and breakdown
[34][122]. Osteocytes are the most prevalent long-lived cell type in bone matrix and are in charge of maintenance of bone mass
[35][123]. Skeletal aging is characterized by bone mass loss and is a significant risk factor for osteoporosis because it results from an increase in osteoclasts and a decrease in osteoblasts count
[36][37][38][39][124,125,126,127]. Cellular senescence has been linked to bone aging and the development of aging-related osteo-pathologies
[40][128]. More precisely, senescent osteocytes have been detected in aging bones with increased expression of p16.
In contrast to osteocytes, senescent osteoblasts are characterized by increased expression of p21 only
[35][38][41][42][43][123,126,129,130,131]. Moreover, the selective elimination of p16-expressing cells using INK-ATTACK transgene resulted in increased bone mass in 20 months old mice
[44][132]. Furthermore, using the p16-3MR transgene, which is based on the elimination of P16-expressing cells upon treatment with ganciclovir (GCV), has effectively abrogated age-related increases in osteoclastogenesis of the myeloid lineage but had no effect on bone formation. This might indicate that p16, rather than direct targeting of senescent osteocytes, contributes to osteoclastogenic potential without major impact on age-related bone loss
[45][133].
However, other implications of p16 have been demonstrated in bone. P16 degradation by the ubiquitinated regulator UBE2S is an important step in the progression of prostate cancer bone metastasis
[46][134]. Furthermore, patients with p16-positive oropharyngeal squamous cell carcinoma had a higher incidence of bone metastasis than p16-negative patients
[47][135]. Lower expression of p16 in osteosarcoma patients was correlated with reduced response to primary chemotherapy
[48][136], which, therefore, shows the importance of p16 as a prognostic and predictive biomarker and therapeutic target for cancer and metastasis.
Aside from p16 in cancer, although only p21-positive cells were able to prevent radiation-induced osteoporosis
[49][86], p16 deletion inhibited oxidative stress, osteocyte senescence, and osteoclastic bone resorption, which led to osteogenesis and osteoblastic bone formation, indicating a promising mechanism to prevent estrogen deficiency-induced osteoporosis
[50][137]. Furthermore, p16 deletion promoted migration, proliferation, and differentiation of bone marrow mesenchymal stem cells (BM-MSCs) and chondrocytes. It also stimulated osteoblastogenesis and vascularization, which improved bone fracture healing. Consequently, p16 modification might offer a novel strategy for treating fractured bones in elderly patients
[51][138] (
Figure 23).
4. In the Lungs
Cellular senescence and aging have both been linked to increased lung damage and functional impairment
[52][139]. Growing evidence suggested aging as another determinant of the chronic obstructive pulmonary disease (COPD) and showed higher prevalence of the disease in elderly
[53][54][55][140,141,142]. Similarly, even though there are no certain causes of idiopathic pulmonary fibrosis (IPF), aging associated with cellular senescence and p16 overexpression has emerged as a main risk factor
[56][57][143,144].
Cigarette smoking (CS) is a major risk factor attributed to COPD
[58][145]. CS can alter cellular proliferation and induce apoptosis, reactive oxygen species production, and promote oxidative stress, cause DNA damage, and trigger cellular senescence
[59][60][146,147]. Furthermore, mice exposed to chronic cigarette smoking at both young and old ages showed increased activation of the senescence marker beta-galactosidase as well as upregulation of p16 compared to their respective air-exposed controls. Older air-exposed mice had higher levels of beta-galactosidase and p16 than younger mice. Therefore, CS-induced senescence and natural-aging-associated senescence are both affected by the p16 pathway
[61][148]. This was confirmed in human COPD patients who had higher p16 expression compared to normal smokers and nonsmokers
[62][149]. Furthermore, after CS exposure, wild type mice had more senescent alveolar type II (AECII) epithelial cells than p16 knockout mice, which had normal pulmonary function. Moreover, p16 deletion has rescued the adverse effects induced by CS in the lungs via the insulin growth factor1 (IGF1)/Akt1 signaling pathway
[62][149].
However, p16 expression is a differentiation key between cervical squamous cell carcinoma (SCC) with pulmonary metastasis and pulmonary SCC. Immunohistochemistry of both cervical SCC without and with pulmonary metastasis has shown an intense staining of p16 in almost all cases studied. On the contrary, cases with pulmonary SCC demonstrated p16 expression in 7 out of 33 cases, 3 of which showed weak p16 staining. This implies the usefulness of p16 as a distinguishing marker between cervical SCC with lung metastasis and pulmonary SCC
[63][150]. Furthermore, the fact that aberrant p16 methylation occurs at early stages of lung cancer renders p16 an early diagnostic biomarker for monitoring and prevention
[64][151]. Moreover, p16 low expression and gene mutation were associated with early and late stage nonsmall cells lung carcinoma (NSCLC), respectively
[65][66][152,153]. As a result, it has been identified as a predictable prognostic factor in NSCLC, particularly at the early stage.
On the other hand, p16 expression is not only linked with disease progression but also with lung protection. P16 loss was linked with poor survival after lung injury. In addition, p16 expression was found to be crucial for protection of lung epithelium against oncogenic stress and lung injury
[67][154]. Moreover, injured p16-positive mesenchymal cells enhanced epithelial progenitor proliferation, whereas deletion of p16 attenuated normal epithelial repair in the lungs
[68][155]. Furthermore, prevalent usefulness was demonstrated for p16 as a target for COPD therapy. Higher p16 expression was found in human COPD lungs compared to normal patients, and when CS induced impaired pulmonary function and augmented emphysema in WT mice, p16 knockout mice exhibited normal pulmonary function with reduced emphysema and increased alveolar progenitor proliferation
[62][149] (
Figure 23).
5. In the Brain
Aging-induced p16 overexpression and cellular senescence have been linked to decreased subventricular zone progenitor proliferation and neurogenesis of the olfactory bulb and to diminished multipotent progenitor cell frequency and self-renewal potency
[69][156]. Moreover, chronic accumulation of senescent cells and the resulting inflammation in the brain has been linked to the development of Alzheimer’s disease (AD) and other neurodegenerative diseases
[70][71][157,158]. In two out of five AD models, Dorigatti et al.
[72][159] found evidence of cellular senescence marked by a significant increase in p16, p21, and p53 expression, as well as increased SASPs expression and beta-galactosidase activity
[72][159]. Another study found that tau-containing neurofibrillary tangles (NFTs), a hallmark of Alzheimer’s disease, are age-dependent and strongly associated with senescence induction and upregulation of p16 and p21
[73][74][160,161]. Nonetheless, astrocytes play an important role in neuronal homeostasis and functions, and, as we age, they undergo senescence in response to multiple stresses, resulting in impaired brain function
[75][76][77][78][162,163,164,165].
As previously discussed for other tumors, unsurprisingly, p16 homozygous deletion was found in both primary glioblastoma and their derived xenografts
[79][167]. In addition, p16-cdk4/cyclin D1-pRb pathway inactivation was found in the majority of glioblastomas
[80][168]. P16 loss was linked to significantly poor outcome in all glioma patients, which indicates a predictive prognostic usefulness of p16 in brain tumors
[81][169]. On the contrary, p16 null glioma cells demonstrated higher chemosensitivity to paclitaxel and topotecan compared to exogenous wild type p16 overexpression
[82][170].
P16 overexpression has been shown to exert a protective function of neurons against CDK overexpression-induced apoptosis
[83][171]. Moreover, increased expression of p16 and p21, induced by stress conditions, has protected female but not male astrocytes from transformation
[84][172]. In another promising strategy, the selective elimination of p16-positive senescent astrocytes diminished cognitive impairment induced by whole brain irradiation
[85][173]. Lastly, dihydromyricetin (DMY), through the downregulation of p16, p21, and p53, was able to inhibit oxidative stress and neuroinflammation and to attenuate brain aging and improve cognitive function in mice
[86][174] (
Figure 23).
6. In the Heart
Remarkable p16 expression and cellular senescence were found in cardiac chronological aging and heart failure
[87][88][175,176]. For example, elevated p16 expression and beta-galactosidase activity were found in cardiomyocytes gathered from Langendorff heart perfusion with aging
[89][91]. In addition to that, cardiac progenitor cells isolated from elderly (> 70 years old) people expressed high levels of p16 and SASPs, alongside shortened telomeres and increased SA-β-gal
[90][92]. Furthermore, remarkable telomere shortening and senescent-associated increased p16 expression were found in cardiomyocytes isolated from old rats compared to younger ones
[91][177]. Older patients with heart failure had higher p16 expression, which was associated with senescence and cell death, as well as shorter telomere length, when compared to healthy elderly people. This suggests that p16-induced senescence, telomere attrition, and cell death are features of heart failure in aging
[88][176]. Furthermore, vascular smooth muscle cell (VSMC) senescence in atherosclerotic plaques was marked by increased p16, p21, and p53 expression in addition to increased beta-galactosidase activity
[92][178].
The recovery of cardiac function and cardiac remodeling have been correlated with cardiac stem cell (CSCs) regeneration and differentiation ability
[93][94][179,180]. Cellular senescence has an impact on CSCs and cardiac function, which might provide a concept of therapies by targeting senescent cells for cardiac functional improvement and extended lifespan in elderly people
[93][179]. With aging, a significant portion of human CSCs become senescent with elevated expression of p16, SA-β-gal, and SASPs, which contribute to CSC senescence and impaired cardiac regeneration. However, INK-ATTAC or senolytic elimination of senescent CSCs reactivated resident CSCs and increased cardiomyocyte proliferation
[93][179] reflecting the importance of p16-positive senescent CSCs as therapeutic approach for cardiac functional improvement. P16-positive cells that accumulate during adulthood have a negative impact on lifespan and promote age-dependent changes in the heart. The removal of p16-positive cells delayed age-related heart deterioration. Thus, the therapeutic removal of these cells may be an appealing approach to extend healthy lifespan
[95][8].
On the contrary to the previous studies, the existence of p16 high cells detected in p16-CreERT2-tdTomato mouse model, was found indispensable for health span, and their elimination has induced cardiac fibrosis
[96][83]. Furthermore, p16 overexpression has been detected in the infarction zone after myocardial infarction. The increased expression of p16 was associated with protected cardiac function and plays an important role for cardiac remodeling after myocardial infarction
[97][181] (
Figure 23).
7. In the Kidney
Several studies have linked p16 induction and subsequent cellular senescence to renal aging, diseases, and allograft rejection
[98][99][100][182,183,184]. Age-dependent p16 upregulation in cortical tubular and interstitial cells was observed in humans. In addition, p16 and p27 expression were higher in the glomeruli, tubules, and interstitial cells of rejected grafts compared to normal kidneys
[99][183]. Whether this reflects senescence as the underlying mechanism for chronic allograft rejection as suggested or might correspond to reduced proliferation and repair or to an increased immune reaction remains to be determined. In line with this, in human kidney specimens ranging from 8 weeks to 88 years of age, p16 induction was negatively correlated with the proliferation marker Ki-67
[101][185], which is in agreement with the role of p16 as a cell cycle inhibitor. Levels of p16 in glomerular and interstitial cells were significantly higher in kidneys with glomerular disease than in normal aged kidneys and kidneys with tubular interstitial nephritis. P16 expression was higher in kidneys with proteinuria, with fibrosis, or interstitial inflammation
[102][186]. Whether this increased P16 expression is cause or consequence of glomerular disease remains an open question. Similarly, increased p16 expression was observed in kidneys of hypertensive animals and patients and kidneys with type 2 diabetic nephropathy
[103][104][187,188]. Blood pressure lowering reduced p16 expression
[103][187], which argues against a close relation between P16 and irreversible senescence in this model. Increased p16 expression has been reported in acute kidney injury (AKI) and in acute tubular necrosis (ATN)
[105][189]. P16 deletion ameliorated ATN and improved kidney function in animal models
[105][189]. Similarly, p16 deletion in Bmi-1-deficient mice rescued kidney aging features including function and structure, ameliorated tubulointerstitial fibrosis, and inhibited epithelial mesenchymal transition of renal interstitial fibroblasts
[106][190] (
Figure 23).
8. In the Liver
Although the majority of liver functions appear to be preserved with age, evidence of aging and cellular senescence associated with liver functional decline, reduced regenerative capacity, and diseases are well-documented
[107][108][109][191,192,193]. P16 expression was higher in elderly hepatectomy patients compared to younger ones, and the increased p16 expression was associated with decreased liver regeneration
[110][194]. This is in agreement with the attenuated proliferative response of hepatocytes in old rat liver compared to young animals
[111][195]. P16 upregulation was observed in liver tissue and liver sinusoidal endothelial cells (LSEC) in an aged rat model compared to young animals
[112][196]. The p16 CreERT2 tdTomato mouse model also demonstrated that p16 high cells were detectable in the liver, and that they were enriched with aging. The majority of the P16-positive liver cells found were vascular endothelial, and their removal caused steatohepatitis and perivascular tissue fibrosis
[96][113][83,197]. This is compatible with higher p16 expression level of liver endothelial cells compared to nonendothelial cells demonstrated in
our
recent study
[31][120].
With respect to liver metabolism, the extra copy of p16 carried by the “Super-INK4A/ARF” mouse model prevented the development of glucose intolerance with aging. Instead, increased activation of insulin receptors and high insulin sensitivity were obtained. This reveals a protective role of INK4A/ARF locus against age-induced insulin resistance
[114][198], whereas increased insulin secretion, attenuated insulin sensitivity, and reduced hepatic insulin clearance were observed upon loss of function mutation of the Cdkn2a gene
[115][199]. On the contrary, p16 deficiency improved fasting-activated glucose production in the liver, via the activation of PKA-CREB-PGC1α
[116][200]. Altogether, these studies show the importance of p16 in glucose homeostasis. However, p16 has not been only implicated in glucose but also in fat metabolism. P16 has been found to regulate fasting-induced fatty acid oxidation and lipid droplet accumulation in the liver in vivo and in vitro. In addition, p16 deficiency was correlated with increased expression of fatty acids catabolism genes in primary hepatocytes
[117][201]. Furthermore, p16-positive senescent cell accumulation has been correlated with hepatic fat deposition and steatosis. Elimination of these cells in the INK-ATTAC mouse model or senolytics treatment (dasatinib plus quercetin) attenuated liver fibrosis
[118][88]. However, the feedback loop between lipid accumulation and increased p16 expression remains intriguing. Senescence in hepatocytes triggered fat accumulation
[118][88], while high fat diet provoked significantly elevated p16 expression
[119][202].
Nonetheless, several studies have also described p16 functions in liver cancers. P16 hypermethylation and consequent p16 inactivation has a pivotal role in the development of hepatocellular carcinoma and liver cirrhosis
[120][205]. Wong et al. reported aberrantly methylated p16 in the plasma of liver cancer patients, suggesting the usefulness of these circulating liver-cancer-methylated DNA for the monitoring of tumors
[121][206]. Therefore, all this information combined suggests that p16 regulation and meticulously unravelling the molecular mechanisms regulating p16 expression in liver physiology and liver pathologies require further elucidation and could unveil novel therapeutic strategies for maintaining normal liver function and extending lifespan (
Figure 23).