 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Agenor Limon | -- | 2485 | 2022-08-31 03:30:38 | | | |
| 2 | Lindsay Dong | -34 word(s) | 2451 | 2022-08-31 03:49:44 | | |
Video Upload Options
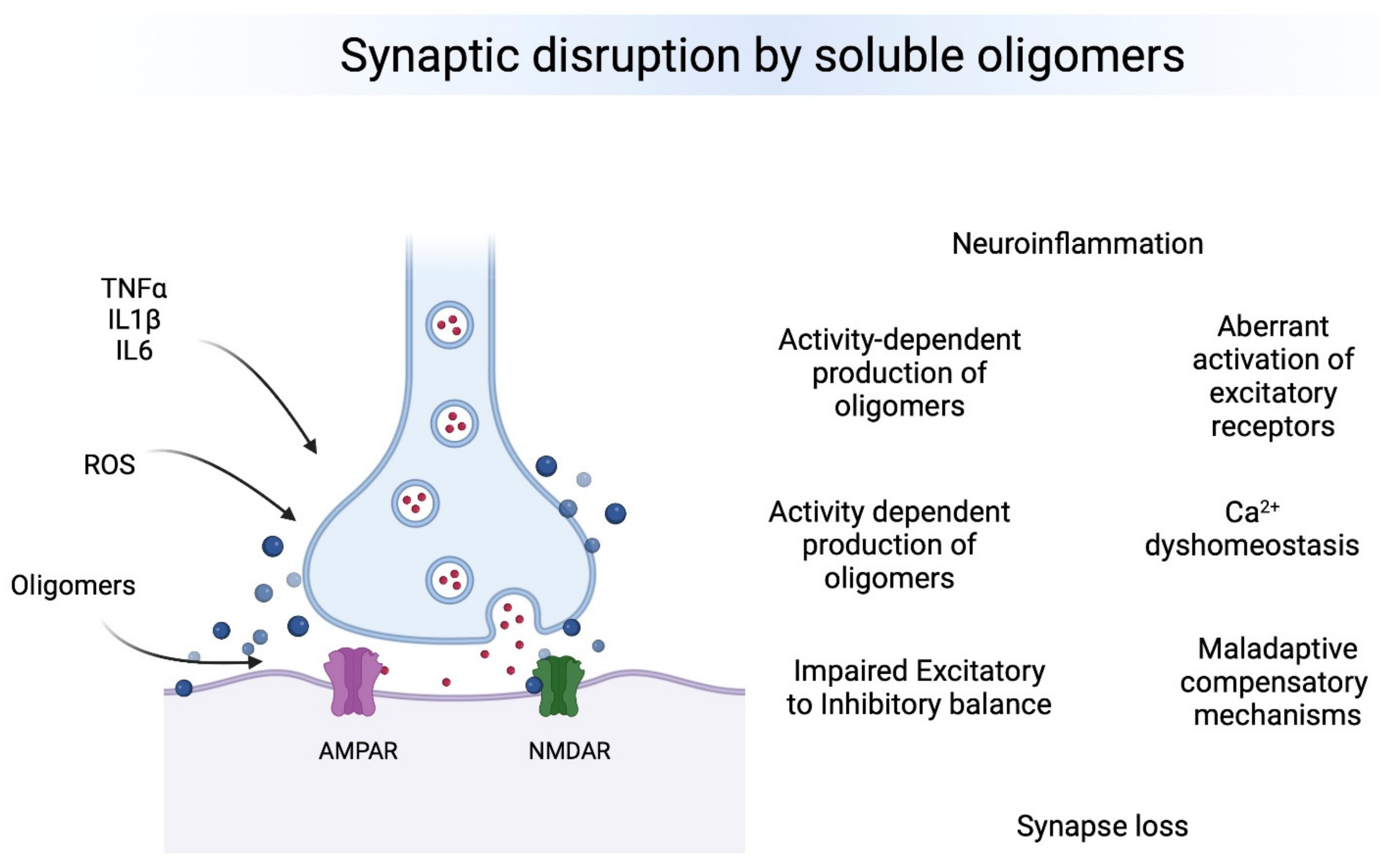
Neurodegenerative diseases are the result of progressive dysfunction of the neuronal activity and subsequent neuronal death. Currently, the most prevalent neurodegenerative diseases are by far Alzheimer’s (AD) and Parkinson’s (PD) disease, affecting millions of people worldwide. Although amyloid plaques and neurofibrillary tangles are the neuropathological hallmarks for AD and Lewy bodies (LB) are the hallmark for PD, current evidence strongly suggests that oligomers seeding the neuropathological hallmarks are more toxic and disease-relevant in both pathologies. The presence of small soluble oligomers is the common bond between AD and PD: amyloid β oligomers (AβOs) and Tau oligomers (TauOs) in AD and α-synuclein oligomers (αSynOs) in PD. Such oligomers appear to be particularly increased during the early pathological stages, targeting synapses at vulnerable brain regions leading to synaptic plasticity disruption, synapse loss, inflammation, excitation to inhibition imbalance and cognitive impairment. Absence of TauOs at synapses in individuals with strong AD disease pathology but preserved cognition suggests that mechanisms of resilience may be dependent on the interactions between soluble oligomers and their synaptic targets.
1. Introduction
2. Neurodegeneration Driven by Small Soluble Oligomers
3. Synaptic Dysfunction Leading to Cognitive Impairment
3.1. Synapse Loss
3.2. Inflammatory Response Effects on Synapses
3.3. Receptors Involved in Synaptic Dysfunction
3.4. Impaired Excitatory/Inhibitory Ratio
4. Conclusions
References
- World Health Organization. Available online: https://www.who.int/news-room/fact-sheets/detail/dementia (accessed on 21 June 2022).
- Parkinson’s Disease Statistics. Available online: https://parkinsonsnewstoday.com/parkinsons-disease-statistics/ (accessed on 5 June 2022).
- Verma, M.; Vats, A.; Taneja, V. Toxic Species in Amyloid Disorders: Oligomers or Mature Fibrils. Ann. Indian Acad. Neurol. 2015, 18, 138–145.
- Forloni, G.; Artuso, V.; la Vitola, P.; Balducci, C. Oligomeropathies and Pathogenesis of Alzheimer and Parkinson’s Diseases. Mov. Disord. 2016, 31, 771–781.
- Ono, K. The Oligomer Hypothesis in α-Synucleinopathy. Neurochem. Res. 2017, 42, 3362–3371.
- Kuo, Y.M.; Emmerling, M.R.; Vigo-Pelfrey, C.; Kasunic, T.C.; Kirkpatrick, J.B.; Murdoch, G.H.; Ball, M.J.; Roher, A.E. Water-Soluble Aβ (N-40, N-42) Oligomers in Normal and Alzheimer Disease Brains. J. Biol. Chem. 1996, 271, 4077–4081.
- Mucke, L.; Selkoe, D.J. Neurotoxicity of Amyloid β-Protein: Synaptic and Network Dysfunction. Cold Spring Harb. Perspect. Med. 2012, 2, a006338.
- Walsh, D.M.; Klyubin, I.; Fadeeva, J.V.; Cullen, W.K.; Anwyl, R.; Wolfe, M.S.; Rowan, M.J.; Selkoe, D.J. Naturally Secreted Oligomers of Amyloid β Protein Potently Inhibit Hippocampal Long-Term Potentiation in Vivo. Nature 2002, 416, 535–539.
- Balducci, C.; Beeg, M.; Stravalaci, M.; Bastone, A.; Sclip, A.; Biasini, E.; Tapella, L.; Colombo, L.; Manzoni, C.; Borsello, T.; et al. Synthetic Amyloid-β Oligomers Impair Long-Term Memory Independently of Cellular Prion Protein. Proc. Natl. Acad. Sci. USA 2010, 107, 2295–2300.
- Haass, C.; Selkoe, D.J. Soluble Protein Oligomers in Neurodegeneration: Lessons from the Alzheimer’s Amyloid β-Peptide. Nat. Rev. Mol. Cell Biol. 2007, 8, 101–112.
- Wilcox, K.C.; Lacor, P.N.; Pitt, J.; Klein, W.L. Aβ Oligomer-Induced Synapse Degeneration in Alzheimer’s Disease. Cell. Mol. Neurobiol. 2011, 31, 939–948.
- Ward, S.M.; Himmelstein, D.S.; Lancia, J.K.; Binder, L.I. Tau Oligomers and Tau Toxicity in Neurodegenerative Disease. Biochem. Soc. Trans. 2012, 40, 667–671.
- Kayed, R.; Lasagna-Reeves, C.A. Molecular Mechanisms of Amyloid Oligomers Toxicity. J. Alzheimer’s Dis. 2013, 33 (Suppl. 1), S67–S78.
- Shankar, G.M.; Li, S.; Mehta, T.H.; Garcia-Munoz, A.; Shepardson, N.E.; Smith, I.; Brett, F.M.; Farrell, M.A.; Rowan, M.J.; Lemere, C.A.; et al. Amyloid-β Protein Dimers Isolated Directly from Alzheimer’s Brains Impair Synaptic Plasticity and Memory. Nat. Med. 2008, 14, 837–842.
- Lacor, P.N.; Buniel, M.C.; Furlow, P.W.; Clemente, A.S.; Velasco, P.T.; Wood, M.; Viola, K.L.; Klein, W.L. Aβ Oligomer-Induced Aberrations in Synapse Composition, Shape, and Density Provide a Molecular Basis for Loss of Connectivity in Alzheimer’s Disease. J. Neurosci. 2007, 27, 796–807.
- Pham, E.; Crews, L.; Ubhi, K.; Hansen, L.; Adame, A.; Cartier, A.; Salmon, D.; Galasko, D.; Michael, S.; Savas, J.N.; et al. Progressive Accumulation of Amyloid-β Oligomers in Alzheimer’s Disease and in Amyloid Precursor Protein Transgenic Mice Is Accompanied by Selective Alterations in Synaptic Scaffold Proteins. FEBS J. 2010, 277, 3051–3067.
- Lesné, S.E.; Sherman, M.A.; Grant, M.; Kuskowski, M.; Schneider, J.A.; Bennett, D.A.; Ashe, K.H. Brain Amyloid-β Oligomers in Ageing and Alzheimer’s Disease. Brain 2013, 136, 1383–1398.
- Lesné, S.; Koh, M.T.; Kotilinek, L.; Kayed, R.; Glabe, C.G.; Yang, A.; Gallagher, M.; Ashe, K.H. A Specific Amyloid-β Protein Assembly in the Brain Impairs Memory. Nature 2006, 440, 352–357.
- Meng, X.; Li, T.; Wang, X.; Lv, X.; Sun, Z.; Zhang, J.; Su, F.; Kang, S.; Kim, S.; An, S.S.A.; et al. Association between Increased Levels of Amyloid-β Oligomers in Plasma and Episodic Memory Loss in Alzheimer’s Disease. Alzheimer’s Res. Ther. 2019, 11, 89.
- McLean, C.A.; Cherny, R.A.; Fraser, F.W.; Fuller, S.J.; Smith, M.J.; Beyreuther, K.; Bush, A.I.; Masters, C.L. Soluble Pool of Aβ Amyloid as a Determinant of Severity of Neurodegeneration in Alzheimer’s Disease. Ann. Neurol. 1999, 46, 860–866.
- Mc Donald, J.M.; Savva, G.M.; Brayne, C.; Welzel, A.T.; Forster, G.; Shankar, G.M.; Selkoe, D.J.; Ince, P.G.; Walsh, D.M. The Presence of Sodium Dodecyl Sulphate-Stable Aβ Dimers Is Strongly Associated with Alzheimer-Type Dementia. Brain 2010, 133, 1328–1341.
- Maeda, S.; Sahara, N.; Saito, Y.; Murayama, S.; Ikai, A.; Takashima, A. Increased Levels of Granular Tau Oligomers: An Early Sign of Brain Aging and Alzheimer’s Disease. Neurosci. Res. 2006, 54, 197–201.
- Gaikwad, S.; Puangmalai, N.; Bittar, A.; Montalbano, M.; Garcia, S.; McAllen, S.; Bhatt, N.; Sonawane, M.; Sengupta, U.; Kayed, R. Tau Oligomer Induced HMGB1 Release Contributes to Cellular Senescence and Neuropathology Linked to Alzheimer’s Disease and Frontotemporal Dementia. Cell Rep. 2021, 36, 109419.
- Patterson, K.R.; Remmers, C.; Fu, Y.; Brooker, S.; Kanaan, N.M.; Vana, L.; Ward, S.; Reyes, J.F.; Philibert, K.; Glucksman, M.J.; et al. Characterization of Prefibrillar Tau Oligomers in Vitro and in Alzheimer Disease. J. Biol. Chem. 2011, 286, 23063–23076.
- Lasagna-Reeves, C.A.; Castillo-Carranza, D.L.; Sengupta, U.; Guerrero-Munoz, M.J.; Kiritoshi, T.; Neugebauer, V.; Jackson, G.R.; Kayed, R. Alzheimer Brain-Derived Tau Oligomers Propagate Pathology from Endogenous Tau. Sci. Rep. 2012, 2, 700.
- Paleologou, K.E.; Kragh, C.L.; Mann, D.M.A.; Salem, S.A.; Al-Shami, R.; Allsop, D.; Hassan, A.H.; Jensen, P.H.; El-Agnaf, O.M.A. Detection of Elevated Levels of Soluble α-Synuclein Oligomers in Post-Mortem Brain Extracts from Patients with Dementia with Lewy Bodies. Brain 2009, 132, 1093–1101.
- Gerson, J.E.; Farmer, K.M.; Henson, N.; Castillo-Carranza, D.L.; Carretero Murillo, M.; Sengupta, U.; Barrett, A.; Kayed, R. Tau Oligomers Mediate α-Synuclein Toxicity and Can Be Targeted by Immunotherapy. Mol. Neurodegener. 2018, 13, 13.
- Surguchev, A.; Surguchov, A. Effect of α-Synuclein on Membrane Permeability and Synaptic Transmission: A Clue to Neurodegeneration? J. Neurochem. 2015, 132, 619–621.
- Winner, B.; Jappelli, R.; Maji, S.K.; Desplats, P.A.; Boyer, L.; Aigner, S.; Hetzer, C.; Loher, T.; Vilar, M.; Campioni, S.; et al. In Vivo Demonstration That α-Synuclein Oligomers Are Toxic. Proc. Natl. Acad. Sci. USA 2011, 108, 4194–4199.
- Surmeier, D.J.; Schumacker, P.T.; Guzman, J.D.; Ilijic, E.; Yang, B.; Zampese, E. Calcium and Parkinson’s Disease. Biochem. Biophys. Res. Commun. 2017, 483, 1013–1019.
- Subramaniam, S.R.; Chesselet, M.-F. Mitochondrial Dysfunction and Oxidative Stress in Parkinson’s Disease. Prog. Neurobiol. 2013, 106–107, 17–32.
- Zampese, E.; Surmeier, D.J. Calcium, Bioenergetics, and Parkinson’s Disease. Cells 2020, 9, 2045.
- Poewe, W.; Seppi, K.; Tanner, C.M.; Halliday, G.M.; Brundin, P.; Volkmann, J.; Schrag, A.-E.; Lang, A.E. Parkinson Disease. Nat. Rev. Dis. Primers 2017, 3, 17013.
- Terry, R.D.; Masliah, E.; Salmon, D.P.; Butters, N.; DeTeresa, R.; Hill, R.; Hansen, L.A.; Katzman, R. Physical Basis of Cognitive Alterations in Alzheimer’s Disease: Synapse Loss Is the Major Correlate of Cognitive Impairment. Ann. Neurol. 1991, 30, 572–580.
- Schulz-Schaeffer, W.J. The Synaptic Pathology of α-Synuclein Aggregation in Dementia with Lewy Bodies, Parkinson’s Disease and Parkinson’s Disease Dementia. Acta Neuropathol. 2010, 120, 131–143.
- Mecca, A.P.; Chen, M.K.; O’Dell, R.S.; Naganawa, M.; Toyonaga, T.; Godek, T.A.; Harris, J.E.; Bartlett, H.H.; Zhao, W.; Nabulsi, N.B.; et al. In Vivo Measurement of Widespread Synaptic Loss in Alzheimer’s Disease with SV2A PET. Alzheimer’s Dement. 2020, 16, 974–982.
- Wilde, M.C.; Overk, C.R.; Sijben, J.W.; Masliah, E. Meta-analysis of Synaptic Pathology in Alzheimer’s Disease Reveals Selective Molecular Vesicular Machinery Vulnerability. Alzheimer’s Dement. 2016, 12, 633–644.
- Matuskey, D.; Tinaz, S.; Wilcox, K.C.; Naganawa, M.; Toyonaga, T.; Dias, M.; Henry, S.; Pittman, B.; Ropchan, J.; Nabulsi, N.; et al. Synaptic Changes in Parkinson Disease Assessed with in Vivo Imaging. Ann. Neurol. 2020, 87, 329–338.
- Forloni, G.; Balducci, C. Alzheimer’s Disease, Oligomers, and Inflammation. J. Alzheimer’s Dis. 2018, 62, 1261–1276.
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s Disease. Lancet Neurol. 2015, 14, 388–405.
- Dahlgren, K.N.; Manelli, A.M.; Stine, W.B.; Baker, L.K.; Krafft, G.A.; LaDu, M.J. Oligomeric and Fibrillar Species of Amyloid-β Peptides Differentially Affect Neuronal Viability. J. Biol. Chem. 2002, 277, 32046–32053.
- Ahmed, M.; Davis, J.; Aucoin, D.; Sato, T.; Ahuja, S.; Aimoto, S.; Elliott, J.I.; van Nostrand, W.E.; Smith, S.O. Structural Conversion of Neurotoxic Amyloid-β1–42 Oligomers to Fibrils. Nat. Struct. Mol. Biol. 2010, 17, 561–567.
- Walsh, D.M.; Selkoe, D.J. Aβ Oligomers—A Decade of Discovery. J. Neurochem. 2007, 101, 1172–1184.
- Michelucci, A.; Heurtaux, T.; Grandbarbe, L.; Morga, E.; Heuschling, P. Characterization of the Microglial Phenotype under Specific Pro-Inflammatory and Anti-Inflammatory Conditions: Effects of Oligomeric and Fibrillar Amyloid-β. J. Neuroimmunol. 2009, 210, 3–12.
- Heurtaux, T.; Michelucci, A.; Losciuto, S.; Gallotti, C.; Felten, P.; Dorban, G.; Grandbarbe, L.; Morga, E.; Heuschling, P. Microglial Activation Depends on β-Amyloid Conformation: Role of the Formylpeptide Receptor 2. J. Neurochem. 2010, 114, 576–586.
- He, Y.; Zheng, M.-M.; Ma, Y.; Han, X.-J.; Ma, X.-Q.; Qu, C.-Q.; Du, Y.-F. Soluble Oligomers and Fibrillar Species of Amyloid β-Peptide Differentially Affect Cognitive Functions and Hippocampal Inflammatory Response. Biochem. Biophys. Res. Commun. 2012, 429, 125–130.
- Rao, J.S.; Kellom, M.; Kim, H.-W.; Rapoport, S.I.; Reese, E.A. Neuroinflammation and Synaptic Loss. Neurochem. Res. 2012, 37, 903–910.
- Lourenco, M.V.; Clarke, J.R.; Frozza, R.L.; Bomfim, T.R.; Forny-Germano, L.; Batista, A.F.; Sathler, L.B.; Brito-Moreira, J.; Amaral, O.B.; Silva, C.A.; et al. TNF-α Mediates PKR-Dependent Memory Impairment and Brain IRS-1 Inhibition Induced by Alzheimer’s β-Amyloid Oligomers in Mice and Monkeys. Cell Metab. 2013, 18, 831–843.
- Forny-Germano, L.; e Silva, N.L.M.; Batista, A.F.; Brito-Moreira, J.; Gralle, M.; Boehnke, S.E.; Coe, B.C.; Lablans, A.; Marques, S.A.; Martinez, A.M.B.; et al. Alzheimer’s Disease-like Pathology Induced by Amyloid-β Oligomers in Nonhuman Primates. J. Neurosci. 2014, 34, 13629–13643.
- Ledo, J.H.; Azevedo, E.P.; Clarke, J.R.; Ribeiro, F.C.; Figueiredo, C.P.; Foguel, D.; de Felice, F.G.; Ferreira, S.T. Amyloid-β Oligomers Link Depressive-like Behavior and Cognitive Deficits in Mice. Mol. Psychiatry 2013, 18, 1053–1054.
- Xu, H.; Gelyana, E.; Rajsombath, M.; Yang, T.; Li, S.; Selkoe, D. Environmental Enrichment Potently Prevents Microglia-Mediated Neuroinflammation by Human Amyloid β-Protein Oligomers. J. Neurosci. 2016, 36, 9041–9056.
- Ledo, J.H.; Azevedo, E.P.; Beckman, D.; Ribeiro, F.C.; Santos, L.E.; Razolli, D.S.; Kincheski, G.C.; Melo, H.M.; Bellio, M.; Teixeira, A.L.; et al. Cross Talk Between Brain Innate Immunity and Serotonin Signaling Underlies Depressive-Like Behavior Induced by Alzheimer’s Amyloid-β Oligomers in Mice. J. Neurosci. 2016, 36, 12106–12116.
- Balducci, C.; Frasca, A.; Zotti, M.; la Vitola, P.; Mhillaj, E.; Grigoli, E.; Iacobellis, M.; Grandi, F.; Messa, M.; Colombo, L.; et al. Toll-like Receptor 4-Dependent Glial Cell Activation Mediates the Impairment in Memory Establishment Induced by β-Amyloid Oligomers in an Acute Mouse Model of Alzheimer’s Disease. Brain Behav. Immun. 2017, 60, 188–197.
- Balducci, C.; Forloni, G. Doxycycline for Alzheimer’s Disease: Fighting β-Amyloid Oligomers and Neuroinflammation. Front. Pharmacol. 2019, 10, 738.
- Hong, S.; Beja-Glasser, V.F.; Nfonoyim, B.M.; Frouin, A.; Li, S.; Ramakrishnan, S.; Merry, K.M.; Shi, Q.; Rosenthal, A.; Barres, B.A.; et al. Complement and Microglia Mediate Early Synapse Loss in Alzheimer Mouse Models. Science 2016, 352, 712–716.
- Lasagna-Reeves, C.A.; Castillo-Carranza, D.L.; Sengupta, U.; Sarmiento, J.; Troncoso, J.; Jackson, G.R.; Kayed, R. Identification of Oligomers at Early Stages of Tau Aggregation in Alzheimer’s Disease. FASEB J. 2012, 26, 1946–1959.
- Shafiei, S.S.; Guerrero-Muñoz, M.J.; Castillo-Carranza, D.L. Tau Oligomers: Cytotoxicity, Propagation, and Mitochondrial Damage. Front. Aging Neurosci. 2017, 9, 83.
- Nilson, A.N.; English, K.C.; Gerson, J.E.; Barton Whittle, T.; Nicolas Crain, C.; Xue, J.; Sengupta, U.; Castillo-Carranza, D.L.; Zhang, W.; Gupta, P.; et al. Tau Oligomers Associate with Inflammation in the Brain and Retina of Tauopathy Mice and in Neurodegenerative Diseases. J. Alzheimer’s Dis. 2017, 55, 1083–1099.
- Alberdi, E.; Sánchez-Gómez, M.V.; Cavaliere, F.; Pérez-Samartín, A.; Zugaza, J.L.; Trullas, R.; Domercq, M.; Matute, C. Amyloid β Oligomers Induce Ca2+ Dysregulation and Neuronal Death through Activation of Ionotropic Glutamate Receptors. Cell Calcium 2010, 47, 264–272.
- Traynelis, S.F.; Wollmuth, L.P.; McBain, C.J.; Menniti, F.S.; Vance, K.M.; Ogden, K.K.; Hansen, K.B.; Yuan, H.; Myers, S.J.; Dingledine, R. Glutamate Receptor Ion Channels: Structure, Regulation, and Function. Pharmacol. Rev. 2010, 62, 405–496.
- Schwenk, J.; Harmel, N.; Zolles, G.; Bildl, W.; Kulik, A.; Heimrich, B.; Chisaka, O.; Jonas, P.; Schulte, U.; Fakler, B.; et al. Functional Proteomics Identify Cornichon Proteins as Auxiliary Subunits of AMPA Receptors. Science 2009, 323, 1313–1319.
- De Boer, H.; Blok, G.J.; Voerman, H.J.; van der Veen, E.A. Is Growth Hormone Supplementation in Growth Hormone Deficiency in Adults Indicated? Ned. Tijdschr. Geneeskd. 1990, 134, 2428–2431.
- Herring, B.E.; Shi, Y.; Suh, Y.H.; Zheng, C.-Y.; Blankenship, S.M.; Roche, K.W.; Nicoll, R.A. Cornichon Proteins Determine the Subunit Composition of Synaptic AMPA Receptors. Neuron 2013, 77, 1083–1096.
- Kato, A.S.; Gill, M.B.; Yu, H.; Nisenbaum, E.S.; Bredt, D.S. TARPs Differentially Decorate AMPA Receptors to Specify Neuropharmacology. Trends Neurosci. 2010, 33, 241–248.
- Cho, C.-H.; St-Gelais, F.; Zhang, W.; Tomita, S.; Howe, J.R. Two Families of TARP Isoforms That Have Distinct Effects on the Kinetic Properties of AMPA Receptors and Synaptic Currents. Neuron 2007, 55, 890–904.
- Milstein, A.D.; Zhou, W.; Karimzadegan, S.; Bredt, D.S.; Nicoll, R.A. TARP Subtypes Differentially and Dose-Dependently Control Synaptic AMPA Receptor Gating. Neuron 2007, 55, 905–918.
- Noh, K.-M.; Yokota, H.; Mashiko, T.; Castillo, P.E.; Zukin, R.S.; Bennett, M.V.L. Blockade of Calcium-Permeable AMPA Receptors Protects Hippocampal Neurons against Global Ischemia-Induced Death. Proc. Natl. Acad. Sci. USA 2005, 102, 12230–12235.
- Liu, S.; Lau, L.; Wei, J.; Zhu, D.; Zou, S.; Sun, H.-S.; Fu, Y.; Liu, F.; Lu, Y. Expression of Ca2+-Permeable AMPA Receptor Channels Primes Cell Death in Transient Forebrain Ischemia. Neuron 2004, 43, 43–55.
- Spaethling, J.M.; Klein, D.M.; Singh, P.; Meaney, D.F. Calcium-Permeable AMPA Receptors Appear in Cortical Neurons after Traumatic Mechanical Injury and Contribute to Neuronal Fate. J. Neurotrauma 2008, 25, 1207–1216.
- Corona, J.C.; Tapia, R. Ca2+-Permeable AMPA Receptors and Intracellular Ca2+ Determine Motoneuron Vulnerability in Rat Spinal Cord in Vivo. Neuropharmacology 2007, 52, 1219–1228.
- Vieira, M.; Fernandes, J.; Burgeiro, A.; Thomas, G.M.; Huganir, R.L.; Duarte, C.B.; Carvalho, A.L.; Santos, A.E. Excitotoxicity through Ca2+-Permeable AMPA Receptors Requires Ca2+-Dependent JNK Activation. Neurobiol. Dis. 2010, 40, 645–655.
- Reinders, N.R.; Pao, Y.; Renner, M.C.; da Silva-Matos, C.M.; Lodder, T.R.; Malinow, R.; Kessels, H.W. Amyloid-β Effects on Synapses and Memory Require AMPA Receptor Subunit GluA3. Proc. Natl. Acad. Sci. USA 2016, 113, E6526–E6534.
- Berchtold, N.C.; Sabbagh, M.N.; Beach, T.G.; Kim, R.C.; Cribbs, D.H.; Cotman, C.W. Brain Gene Expression Patterns Differentiate Mild Cognitive Impairment from Normal Aged and Alzheimer’s Disease. Neurobiol. Aging 2014, 35, 1961–1972.
- Texidó, L.; Martín-Satué, M.; Alberdi, E.; Solsona, C.; Matute, C. Amyloid β Peptide Oligomers Directly Activate NMDA Receptors. Cell Calcium 2011, 49, 184–190.
- Sinnen, B.L.; Bowen, A.B.; Gibson, E.S.; Kennedy, M.J. Local and Use-Dependent Effects of β-Amyloid Oligomers on NMDA Receptor Function Revealed by Optical Quantal Analysis. J. Neurosci. 2016, 36, 11532–11543.
- Ferreira, I.L.; Bajouco, L.M.; Mota, S.I.; Auberson, Y.P.; Oliveira, C.R.; Rego, A.C. Amyloid β Peptide 1–42 Disturbs Intracellular Calcium Homeostasis through Activation of GluN2B-Containing N-Methyl-d-Aspartate Receptors in Cortical Cultures. Cell Calcium 2012, 51, 95–106.
- Kessels, H.W.; Nabavi, S.; Malinow, R. Metabotropic NMDA Receptor Function Is Required for β-Amyloid-Induced Synaptic Depression. Proc. Natl. Acad. Sci. USA 2013, 110, 4033–4038.
- Snyder, E.M.; Nong, Y.; Almeida, C.G.; Paul, S.; Moran, T.; Choi, E.Y.; Nairn, A.C.; Salter, M.W.; Lombroso, P.J.; Gouras, G.K.; et al. Regulation of NMDA Receptor Trafficking by Amyloid-β. Nat. Neurosci. 2005, 8, 1051–1058.
- Ferreira, I.L.; Ferreiro, E.; Schmidt, J.; Cardoso, J.M.; Pereira, C.M.F.; Carvalho, A.L.; Oliveira, C.R.; Rego, A.C. Aβ and NMDAR Activation Cause Mitochondrial Dysfunction Involving ER Calcium Release. Neurobiol. Aging 2015, 36, 680–692.
- Costa, R.O.; Lacor, P.N.; Ferreira, I.L.; Resende, R.; Auberson, Y.P.; Klein, W.L.; Oliveira, C.R.; Rego, A.C.; Pereira, C.M.F. Endoplasmic Reticulum Stress Occurs Downstream of GluN2B Subunit of N-Methyl-d-Aspartate Receptor in Mature Hippocampal Cultures Treated with Amyloid-β Oligomers. Aging Cell 2012, 11, 823–833.
- Evans, R.C.; Morera-Herreras, T.; Cui, Y.; Du, K.; Sheehan, T.; Kotaleski, J.H.; Venance, L.; Blackwell, K.T. The Effects of NMDA Subunit Composition on Calcium Influx and Spike Timing-Dependent Plasticity in Striatal Medium Spiny Neurons. PLoS Comput. Biol. 2012, 8, e1002493.
- Skeberdis, V.A.; Chevaleyre, V.; Lau, C.G.; Goldberg, J.H.; Pettit, D.L.; Suadicani, S.O.; Lin, Y.; Bennett, M.V.L.; Yuste, R.; Castillo, P.E.; et al. Protein Kinase A Regulates Calcium Permeability of NMDA Receptors. Nat. Neurosci. 2006, 9, 501–510.
- Amatniek, J.C.; Hauser, W.A.; DelCastillo-Castaneda, C.; Jacobs, D.M.; Marder, K.; Bell, K.; Albert, M.; Brandt, J.; Stern, Y. Incidence and Predictors of Seizures in Patients with Alzheimer’s Disease. Epilepsia 2006, 47, 867–872.
- Palop, J.J.; Mucke, L. Epilepsy and Cognitive Impairments in Alzheimer Disease. Arch. Neurol. 2009, 66, 435–440.
- Hauser, W.A.; Morris, M.L.; Heston, L.L.; Anderson, V.E. Seizures and Myoclonus in Patients with Alzheimer’s Disease. Neurology 1986, 36, 1226–1230.
- Mendez, M.; Lim, G. Seizures in Elderly Patients with Dementia: Epidemiology and Management. Drugs Aging 2003, 20, 791–803.
- Jayadev, S.; Leverenz, J.B.; Steinbart, E.; Stahl, J.; Klunk, W.; Yu, C.-E.; Bird, T.D. Alzheimer’s Disease Phenotypes and Genotypes Associated with Mutations in Presenilin 2. Brain 2010, 133, 1143–1154.
- Cabrejo, L.; Guyant-Maréchal, L.; Laquerrière, A.; Vercelletto, M.; de la Fournière, F.; Thomas-Antérion, C.; Verny, C.; Letournel, F.; Pasquier, F.; Vital, A.; et al. Phenotype Associated with APP Duplication in Five Families. Brain 2006, 129, 2966–2976.
- Sperling, R.A.; Laviolette, P.S.; O’Keefe, K.; O’Brien, J.; Rentz, D.M.; Pihlajamaki, M.; Marshall, G.; Hyman, B.T.; Selkoe, D.J.; Hedden, T.; et al. Amyloid Deposition Is Associated with Impaired Default Network Function in Older Persons without Dementia. Neuron 2009, 63, 178–188.
- Palop, J.J.; Chin, J.; Roberson, E.D.; Wang, J.; Thwin, M.T.; Bien-Ly, N.; Yoo, J.; Ho, K.O.; Yu, G.-Q.; Kreitzer, A.; et al. Aberrant Excitatory Neuronal Activity and Compensatory Remodeling of Inhibitory Hippocampal Circuits in Mouse Models of Alzheimer’s Disease. Neuron 2007, 55, 697–711.
- Verret, L.; Mann, E.O.; Hang, G.B.; Barth, A.M.I.; Cobos, I.; Ho, K.; Devidze, N.; Masliah, E.; Kreitzer, A.C.; Mody, I.; et al. Inhibitory Interneuron Deficit Links Altered Network Activity and Cognitive Dysfunction in Alzheimer Model. Cell 2012, 149, 708–721.
- Sos, K.E.; Mayer, M.I.; Takács, V.T.; Major, A.; Bardóczi, Z.; Beres, B.M.; Szeles, T.; Saito, T.; Saido, T.C.; Mody, I.; et al. Amyloid β Induces Interneuron-Specific Changes in the Hippocampus of APPNL-F Mice. PLoS ONE 2020, 15, e0233700.
- Li, G.; Bien-Ly, N.; Andrews-Zwilling, Y.; Xu, Q.; Bernardo, A.; Ring, K.; Halabisky, B.; Deng, C.; Mahley, R.W.; Huang, Y. GABAergic Interneuron Dysfunction Impairs Hippocampal Neurogenesis in Adult Apolipoprotein E4 Knockin Mice. Cell Stem Cell 2009, 5, 634–645.
- Andrews-Zwilling, Y.; Bien-Ly, N.; Xu, Q.; Li, G.; Bernardo, A.; Yoon, S.Y.; Zwilling, D.; Yan, T.X.; Chen, L.; Huang, Y. Apolipoprotein E4 Causes Age- and Tau-Dependent Impairment of GABAergic Interneurons, Leading to Learning and Memory Deficits in Mice. J. Neurosci. 2010, 30, 13707–13717.
- Limon, A.; Reyes-Ruiz, J.M.; Miledi, R. Loss of Functional GABAA Receptors in the Alzheimer Diseased Brain. Proc. Natl. Acad. Sci. USA 2012, 109, 10071–10076.
- Berchtold, N.C.; Coleman, P.D.; Cribbs, D.H.; Rogers, J.; Gillen, D.L.; Cotman, C.W. Synaptic Genes Are Extensively Downregulated across Multiple Brain Regions in Normal Human Aging and Alzheimer’s Disease. Neurobiol. Aging 2013, 34, 1653–1661.

