Accumulated evidence indicates that AβOs directly activates AMPA receptors [
105]. AMPA receptors are complex proteins made by the combination of four principal subunits (GluA1-GluA4) [
105,
106], and co-assembled auxiliary proteins [
107,
108], that modulate the gating, permeability, and pharmacology of the channel [
108,
109,
110,
111,
112]. GluA2-lacking AMPA receptors are permeable to Ca
2+ and its excessive activation leads to Ca
2+ overload, excitotoxicity, and neurodegeneration [
113,
114,
115,
116,
117]. Recent evidence from Reinders et al., demonstrated that AβOs cause synaptic failure only in neurons expressing GluA3 subunits [
118], and mice with severe AD neuropathology but deficient in GluA3 were cognitively resilient [
118], strongly indicating that synaptic vulnerability to AβOs may depend on the stoichiometry of synaptic receptors. This is consistent with human postmortem studies where lower gene expression levels for GluA3 correlated with better cognitive performance in prodromal AD [
119]. Similarly, it is increasingly acknowledged that AβOs directly activate heterologously-expressed receptors composed by GRIN1/GluN2A and GRIN1/GluN2B subunits [
105,
120], which are the most abundant NMDA receptors in mammals’ cortical synapses; however, only the activation of receptors containing GluN2B subunits (GluN2B-NMDA receptors) leads to acute activity-dependent postsynaptic failure [
121], Ca
2+ dysregulation [
122], synaptic depression [
123,
124], and neurotoxicity in in vitro systems [
125,
126]. Most likely due to the high Ca
2+ permeability of GluN2B-NMDA receptors [
127] and their downstream signaling [
128]. The clinical significance of GluN2B is reinforced by a multisite postmortem study showing that lower cortical gene expression of GluN2B correlates with better cognitive performance in people diagnosed with prodromal AD [
119].
3.4. Impaired Excitatory/Inhibitory Ratio
Hyperexcitability of cortical and hippocampal circuits and 87-fold increase in seizures incidence in the AD population is well documented [
135], particularly in early-onset familial AD [
136]. Convulsive seizures occur in approximately 7–21% of sporadic AD patients [
137,
138], 31% of patients with PS2 mutations [
139] and 56% of patients with APP duplications [
140]. These data do not account for hidden hyperexcitability status that occurs early in AD pathogenesis [
141]. Since oligomers act mostly on excitatory synapses leading to dysfunction first and synaptic loss later, a large question in the field is how, overall reduction of excitatory inputs leads to hyperexcitability in the AD brain. Although the causes of network hyperactivity are still under investigation by many labs; early studies in animal models suggest that impaired inhibition is a potential mechanism for network hyperactivity [
142,
143]. Impairment of interneuron activity with changes in their intrinsic properties have been reported in the mice models of amyloidosis [
144]. Interneuron deficits reduces neurogenesis and neuronal maturation in the hilus of the hippocampus [
145] and leads to age and tau-dependent learning and memory deficits [
146]. Potentiation of GABA receptors by pentobarbital restores some of the deficits observed by GABAergic impairment [
145]. Our initial studies transplanting human receptors and recording their electrical activity observed a dramatic reduction of GABA
A receptors in AD [
147]. This severe reduction of gene expression was later confirmed by other groups using high throughput microarray technology [
148] and provided evidence that in addition to excitatory synaptic loss, inhibitory synapses were also affected in AD.
4. Conclusions
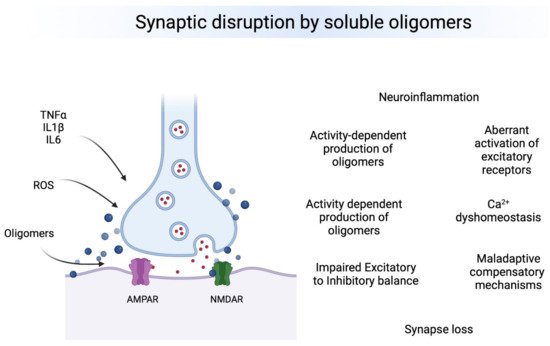
Oligomeric forms of Aβ, tau and aSyn are the most toxic species affecting synapses leading to synaptic dysfunction and altered neuronal communication in brain regions vulnerable to the neuropathology. The effects of oligomers precede the presence of deposits and seem to be associated to early changes in excitatory and inhibitory synapses. Therefore, oligomers seem to produce a “double hit” on synapses (Figure 1). First, they lead to calcium dys-homeostasis by binding directly to excitatory receptors and leading to a first wave of hyperexcitability, then producing GABAergic dysfunction by a mechanism that is still not understood, which leads to a second chronic wave of hyperexcitablity that ultimately leads to neuronal loss and hypoactivity. Understanding the regional and temporal relationships between oligomers, synaptic targets and E/I balance is a critical need in the field.
Figure 1. Overview of major effects of toxic oligomers in synapses. Left. Neuroinflammatory and Reactive Oxygen Species (ROS) participate in the production and the effects of toxic oligomers on synapses. Right, Major synaptic effects on synapses. It is still not clear what is the chronological order of events, but each one influence the others and some of them are happening simultaneously at brain regions vulnerable to AD pathology.