Neurodegenerative diseases are the result of progressive dysfunction of the neuronal activity and subsequent neuronal death. Currently, the most prevalent neurodegenerative diseases are by far Alzheimer’s (AD) and Parkinson’s (PD) disease, affecting millions of people worldwide. Although amyloid plaques and neurofibrillary tangles are the neuropathological hallmarks for AD and Lewy bodies (LB) are the hallmark for PD, current evidence strongly suggests that oligomers seeding the neuropathological hallmarks are more toxic and disease-relevant in both pathologies. The presence of small soluble oligomers is the common bond between AD and PD: amyloid β oligomers (AβOs) and Tau oligomers (TauOs) in AD and α-synuclein oligomers (αSynOs) in PD. Such oligomers appear to be particularly increased during the early pathological stages, targeting synapses at vulnerable brain regions leading to synaptic plasticity disruption, synapse loss, inflammation, excitation to inhibition imbalance and cognitive impairment. Absence of TauOs at synapses in individuals with strong AD disease pathology but preserved cognition suggests that mechanisms of resilience may be dependent on the interactions between soluble oligomers and their synaptic targets.
- oligomers
- E/I balance
- neurodegenerative diseases
1. Introduction
2. Neurodegeneration Driven by Small Soluble Oligomers
Oligomers are small, soluble protein aggregates which possess unique structural and functional properties. They are intermediary between soluble monomeric proteins and insoluble mature fibrils [3][26]. For several years, therapeutic research in AD and PD was centered in targeting insoluble fibrillar aggregates of Aβ, tau and αSyn, but recent studies have shown that soluble oligomers are the most toxic species that induce neuronal damage and dysfunction in neurodegenerative disorders [4][5][6][7][8][9][10][11][12][13][27,28,29,30,31,32,33,34,35,36], suggesting that anti-oligomeric therapeutic strategies would be a better approach to antagonize cognitive deficit symptoms. Neurodegeneration driven by AβOs has been experimentally proven using human postmortem brain tissue from subjects clinically diagnosed with AD. AβOs extracted from these subjects have shown to alter long-term potentiation, enhance long term depression, and reduce the dendritic spine density of pyramidal neurons in the hippocampus of control mice [14][37]. The reduction of spine density is consequence of the loss of spine cytoskeletal proteins, a phenomenon that implicates impairment of memory-related receptors such as NMDA receptors [14][15][37,38] (See below for effects of oligomers on synaptic receptors). AβOs also contribute to loss of synaptic markers such as synaptic vesicle-associated membrane protein 2 and post-synaptic density protein 95 [16][39] indicating reduction of synapses and loss of communication between neurons. The synaptic deterioration manifests with memory and learning impairment as observed in control rats after injection of AβOs from human AD brains [14][37]. Further, levels of AβOs in fractionated brain homogenates from patients with AD correlate with the severity of cognitive impairment (assessed by Blessed Information-Memory-Concentration and the Mini-Mental State Examination scores) [16][39]. Nevertheless, AβOs are found in subjects without cognitive impairment; they increase physiologically in old age [17][40]. The fact that both demented and non-demented patients have increased levels of oligomers does not necessarily mean they have the same oligomeric structure. AβOs organize into dimers, trimers, tetramers, and higher order structures. [17][18][40,41]. The relation between oligomer formation and disease state remains controversial. Most studies support the pathogenic role of oligomers in neurodegeneration as mentioned above. For example, elevated levels of plasma AβOs have shown a strong correlation with the cognitive performance in patients with AD (assessed by Mini-Mental State Examination, Cognitive Abilities Screening Instrument, Alzheimer’s Disease Assessment Scale–cognitive portion, and common objects memory test) [19][44]. Other studies have shown correlation with severity [20][21][45,46]. TauOs are present in neurons and astrocytes at early stages of neurodegeneration, they are increased even before the formation of neurofibrillary tangles and clinical manifestations of AD [22][23][24][50,51,52]. As with AβOs, human brain derived TauOs injected in mice impair synaptic plasticity in the hippocampus, and clinically manifest with anterograde memory storage dysfunction [25][53]. TauOs are not only increased in senile and AD brains, but also, they have been detected in high levels in animal models and brains of individuals with PD, suggesting that TauOs are neurotoxic mediators in synucleinopathies [26][27][54,55]. Neurodegeneration driven by αSynOs depends on their interaction with cell membranes. Similar to AβOs and TauOs, αSynOs can perforate the membrane and hence alter the membrane conductance; therefore, formation of ion-permeating pores seems to be a general mechanism to destabilize the cell membrane shared by some oligomeric forms of misfolded proteins. Moreover, perfusion of αSynOs onto hippocampal neurons induce an increase of intracellular calcium level, which supports the idea of strong membrane interactions [28][29][60,61]. The elevation of calcium levels is in accordance with the calcium homeostasis dysregulation observed in PD subjects [30][31][32][33][62,63,64,65].3. Synaptic Dysfunction Leading to Cognitive Impairment
3.1. Synapse Loss
It has been well established that synaptic dysfunction occurs in AD and PD. Less synapses are found in postmortem brains of patients with AD and PD in brain regions underlying clinical manifestations of both diseases [34][35][67,68]. However, only recently it became possible to evaluate synaptic alterations in alive people by using synaptic positron emission tomography (PET) imaging. Specifically, the PET tracer [11C]UCB-J for the synaptic vesicle glycoprotein (SV2A), expressed in all synapses and located in synaptic vesicles at presynaptic terminals, was recently used to detect synaptic alterations in vivo of patients with early AD and PD. In AD, PET imaging of SV2A showed prominent reduction synapses in the hippocampus, followed by the entorhinal cortex, parahippocampal cortex, amygdala, lateral temporal cortex, PCC/precuneus, and lateral parietal cortex, but not in the prefrontal cortex, lateral occipital cortex, medial occipital cortex, or pericentral cortex [36][69]. The synaptic density reductions were maintained after partial volume correction of the PET images, meaning that the effect is not entirely attributed to loss of gray matter tissue. Importantly, there was a correlation between the reduction of SV2A and cognitive impairment. PET studies correlate with accumulated literature that has consistently shown evidence of synaptic loss across brain regions in AD and other neurodegenerative disorders [37][70]. In PD, PET imaging showed lower SV2A in the substantia nigra, followed by red nucleus and locus coeruleus as well as other clinically relevant areas [38][71].3.2. Inflammatory Response Effects on Synapses
Although AD and PD were not originally considered inflammatory disorders, neuroinflammation is a critical component in the pathogenesis and progression of cognitive impairment. Neuroinflammation involves activation of microglia and astrocytes, and the subsequent release of cytokine radicals which lead to synaptic loss and damage [39][40][72,73]. Particularly, microglia are pivotal in the control of synapse activity by establishing direct contact with neurons, meaning that an inflammatory process at this level has a negative impact on synaptic surveillance and thus, cognitive function. However, whether neuroinflammation is caused by soluble oligomers, the most toxic components in the pathology of AD and PD, is not clear yet. Distinct Aβ conformations seem to trigger different magnitudes of microglial activation. As mentioned before, oligomeric (rather than fibrillary) forms of Aβ, are the most neurotoxic aggregates in AD [14][15][41][42][43][37,38,74,75,76]. Thus, it has been investigated in vitro and in vivo whether AβOs are also stronger promoters of glial activation. An in vitro glial cell culture exposed to AβOs and fibrillar-Aβ, demonstrated not only that the pro-inflammatory response of the oligomeric form of Aβ was stronger than its fibrillary counterpart, but also that the response was an M1-like phenotype [44][77]. Complementarily, a murine study, where brain inflammation was induced by different Aβ42 conformers, showed that the lightest AβOs can activate microglial cells and promote a violent inflammatory response, whereas heavier oligomeric and fibrillary Aβ conformations induced less glial activation and poorer inflammatory responses [45][78]. Another in vivo model demonstrated AβOs promoted stronger neurotoxicity and inflammatory response mediated by NF-κB, when compared to fibrillar-Aβ [46][79]. All these studies reinforce the idea that AβOs are the most potent activators of microglial cells, and following studies display how this inflammatory response leads to synaptic disruption and sequential neuronal dysfunction. The inflammatory response followed by synaptic disruption and neuronal loss can be clinically translated as memory, language, and visual perception decline, among other forms of cognitive impairment [47][80]. In animal models AβOs induce inflammatory signaling leading to this cognitive decline manifestations [48][49][50][81,82,83]. For instance, in an acute experimental model in C57BL/6 mice, memory impairment and inflammation were observed after an intracerebroventricular injection of AβOs, suggesting that oligomers interfere with synaptic transmission necessary to establish new memories; again, the fibrillar-Aβ did not produce this effect [9][32]. The molecular link between cognitive deficit and neuroinflammation lies in the release of cytokines by microglial cells. In one study, purified AβOs from human AD brain tissue were injected in wild type mice to induce microglial inflammation. The inflammatory response of this model was demonstrated when several cytokines at mRNA and protein levels were identified, including Ccl3, CCl4, and Tnf [51][84]. Other mechanism underlying AβO-induced microglial activation is explained by TLR-4, which likely induces aberrant TNF-α signaling [52][53][85,86]. In support of this deleterious role of the inflammatory response, the cognitive decline, induced by the intracerebral injection of AβOs, was reversed by the administration of anti-inflammatory drugs, doxycycline, and TLR-4 antagonists [54][87]. In another study of intracerebroventricular injection of AβOs in wild type mice, the complement factors C1q, which initiates the classic complement pathway, and C3, were found elevated at the synapse level, which would explain the synapse loss through microglial activation [55][88]. In addition to inflammation induced by microglia, astrocytosis is another early phenomenon in AD development, but whether AβOs induce astrocytosis remains to be determined. TauOs are the most neurotoxic tau species involved in the development of cognitive impairment [56][57][89,90]. They induce neuroinflammation in AD and frontotemporal lobar dementia through interaction with astrocytes and microglia [58][91]. A model for the toxic relationship between TauOs and inflammation has been proposed, where TauOs through astrocytes and microglia trigger the release of cytokines, RAGE receptors and their ligand HMGB1. Activation of RAGE signals NF-κB and p38-MAPK pathways, which in turn promote hyperphosphorylation of Tau and subsequent aggregation of more oligomers, and thus, neuronal damage or death and a vicious cycle of chronic neuroinflammation [23][58][51,91].3.3. Receptors Involved in Synaptic Dysfunction
Accumulated evidence indicates that AβOs directly activates AMPA receptors [59][105]. AMPA receptors are complex proteins made by the combination of four principal subunits (GluA1-GluA4) [59][60][105,106], and co-assembled auxiliary proteins [61][62][107,108], that modulate the gating, permeability, and pharmacology of the channel [62][63][64][65][66][108,109,110,111,112]. GluA2-lacking AMPA receptors are permeable to Ca2+ and its excessive activation leads to Ca2+ overload, excitotoxicity, and neurodegeneration [67][68][69][70][71][113,114,115,116,117]. Recent evidence from Reinders et al., demonstrated that AβOs cause synaptic failure only in neurons expressing GluA3 subunits [72][118], and mice with severe AD neuropathology but deficient in GluA3 were cognitively resilient [72][118], strongly indicating that synaptic vulnerability to AβOs may depend on the stoichiometry of synaptic receptors. This is consistent with human postmortem studies where lower gene expression levels for GluA3 correlated with better cognitive performance in prodromal AD [73][119]. Similarly, it is increasingly acknowledged that AβOs directly activate heterologously-expressed receptors composed by GRIN1/GluN2A and GRIN1/GluN2B subunits [59][74][105,120], which are the most abundant NMDA receptors in mammals’ cortical synapses; however, only the activation of receptors containing GluN2B subunits (GluN2B-NMDA receptors) leads to acute activity-dependent postsynaptic failure [75][121], Ca2+ dysregulation [76][122], synaptic depression [77][78][123,124], and neurotoxicity in in vitro systems [79][80][125,126]. Most likely due to the high Ca2+ permeability of GluN2B-NMDA receptors [81][127] and their downstream signaling [82][128]. The clinical significance of GluN2B is reinforced by a multisite postmortem study showing that lower cortical gene expression of GluN2B correlates with better cognitive performance in people diagnosed with prodromal AD [73][119].3.4. Impaired Excitatory/Inhibitory Ratio
Hyperexcitability of cortical and hippocampal circuits and 87-fold increase in seizures incidence in the AD population is well documented [83][135], particularly in early-onset familial AD [84][136]. Convulsive seizures occur in approximately 7–21% of sporadic AD patients [85][86][137,138], 31% of patients with PS2 mutations [87][139] and 56% of patients with APP duplications [88][140]. These data do not account for hidden hyperexcitability status that occurs early in AD pathogenesis [89][141]. Since oligomers act mostly on excitatory synapses leading to dysfunction first and synaptic loss later, a large question in the field is how, overall reduction of excitatory inputs leads to hyperexcitability in the AD brain. Although the causes of network hyperactivity are still under investigation by many labs; early studies in animal models suggest that impaired inhibition is a potential mechanism for network hyperactivity [90][91][142,143]. Impairment of interneuron activity with changes in their intrinsic properties have been reported in the mice models of amyloidosis [92][144]. Interneuron deficits reduces neurogenesis and neuronal maturation in the hilus of the hippocampus [93][145] and leads to age and tau-dependent learning and memory deficits [94][146]. Potentiation of GABA receptors by pentobarbital restores some of the deficits observed by GABAergic impairment [93][145]. SomeOur initial studies transplanting human receptors and recording their electrical activity observed a dramatic reduction of GABAA receptors in AD [95][147]. This severe reduction of gene expression was later confirmed by other groups using high throughput microarray technology [96][148] and provided evidence that in addition to excitatory synaptic loss, inhibitory synapses were also affected in AD.4. Conclusions
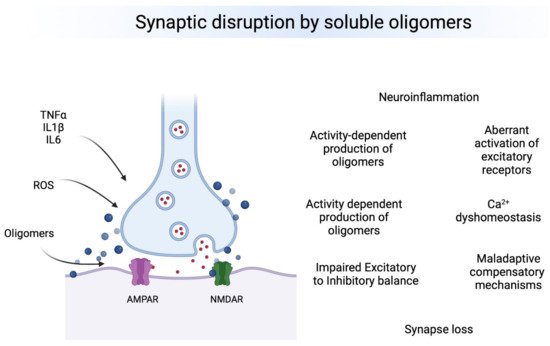
Oligomeric forms of Aβ, tau and aSyn are the most toxic species affecting synapses leading to synaptic dysfunction and altered neuronal communication in brain regions vulnerable to the neuropathology. The effects of oligomers precede the presence of deposits and seem to be associated to early changes in excitatory and inhibitory synapses. Therefore, oligomers seem to produce a “double hit” on synapses (Figure 1). First, they lead to calcium dys-homeostasis by binding directly to excitatory receptors and leading to a first wave of hyperexcitability, then producing GABAergic dysfunction by a mechanism that is still not understood, which leads to a second chronic wave of hyperexcitablity that ultimately leads to neuronal loss and hypoactivity. Understanding the regional and temporal relationships between oligomers, synaptic targets and E/I balance is a critical need in the field.