 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Łukasz Szeleszczuk | + 4477 word(s) | 4477 | 2020-10-13 08:56:18 | | | |
| 2 | Bruce Ren | Meta information modification | 4477 | 2020-10-19 12:00:29 | | |
Video Upload Options
This entry discusses a set of instrumental and computational methods that are used to characterize hydrated forms of APIs (active pharmaceutical ingredients). The focus has been put on highlighting advantages as well as on presenting some limitations of the selected analytical approaches. This has been performed in order to facilitate the choice of an appropriate method depending on the type of the structural feature that is to be analyzed, that is, degree of hydration, crystal structure and dynamics, and (de)hydration kinetics. The presented techniques include X-ray diffraction (single crystal X-ray diffraction (SCXRD), powder X-ray diffraction (PXRD)), spectroscopic (solid state nuclear magnetic resonance spectroscopy (ssNMR), Fourier-transformed infrared spectroscopy (FT-IR), Raman spectroscopy), thermal (differential scanning calorimetry (DSC), thermogravimetric analysis (TGA)), gravimetric (dynamic vapour sorption (DVS)), and computational (molecular mechanics (MM), Quantum Mechanics (QM), molecular dynamics (MD)) methods. Further, the successful applications of the presented methods in the studies of hydrated APIs as well as studies on the excipients’ influence on these processes have been described in many examples.
1. Introduction
Polymorphism is a phenomenon defined as the possibility of one chemical substance to exist in several different crystallographic forms [1] depending on the temperature, pressure, and humidity as well as solvents applied during the crystallization process. Polymorphic forms of active pharmaceutical ingredients (APIs) may differ in certain important properties, such as solubility in water, dissolution rate, melting point, stability, tabletability, and others, which consequently can have an influence on the drug stability and bioavailability [1]. Different group of structures, although in some respects similar to polymorphs, are hydrates. They form a subtype of solid solvates in which water molecules are incorporated into the crystal lattice of a compound [2]. In comparison with related anhydrates, they exhibit a different structure, as it may be altered by the complex H-bonding network [3]. As a consequence, they may also have different physical and chemical properties than their anhydrous counterparts. These parallels to polymorphs are the reasons hydrates were previously called pseudo-polymorphs [4][5]. This naming can be found in old manuscripts, however, nowadays it is no more accepted as the correct one in relation to hydrates. The topic becomes even more complicated when a particular hydrate in itself has different polymorphic forms, for example, nitrofurantoin monohydrate or niclosamide monohydrate [6]. In such cases, the stoichiometry of the system, the number of both host molecules and water molecules, is preserved and only changes in the arrangement of the constituents in the crystal lattice, and alterations in unit cell parameters occur as well.
Hydrates are of particular interest among solid APIs solvates for several reasons. First, the unique character of the water molecule—its relatively small size and the possibility to form the interactions as both a donor and acceptor of H-bonding, sometimes simultaneously, make it an important “building material” in the field of crystal engineering. Further, from the pharmaceutical point of view, it is a non-toxic substance, in contrast to most of the other organic solvents. Finally, owing to the present of the moisture in the air, spontaneous hydration may occur at any stage of drug production or storage, leading to hydrate formation.
Considering the structure, the most common are layer, void, and channel hydrates (Figure 1). They can be also divided into stoichiometric and non-stoichiometric ones. Stoichiometric hydrates are composed of a constant number of water molecules located in clearly defined structure elements like channels [7]. On the contrary, non-stoichiometric hydrates have a variable number of water molecules incorporated in a crystal lattice and, in this case, their naming, for example, dihydrate, usually indicates the maximum number of H2O molecules present in a structure. Non-stoichiometric hydrates can occur as channel or void hydrates, but the solvent molecules are disordered because of the weaker H-bonds with the host molecules, when compared with stoichiometric hydrates [8]. As a result, water diffusion out of these structures is much easier. Furthermore, their hydration level depends highly on the humidity in the surrounding atmosphere. Moreover, non-stoichiometric hydrates are much less prone to collapse after water removal. This phenomenon is characteristic for stoichiometric hydrates, especially when they form large channels or voids. In such cases, distortion of a complex H-bonding network that was stabilizing the stoichiometric hydrate leads to completely new molecular arrangements. Less prevalent among API hydrates are ion coordinated hydrates (known also as ion-associated hydrates) [9] and isolated site hydrates. In case of the latter, water molecules interact solely with the host molecule, but not with each other [10].
|
(a) |
|
(b) |
|
(c) |
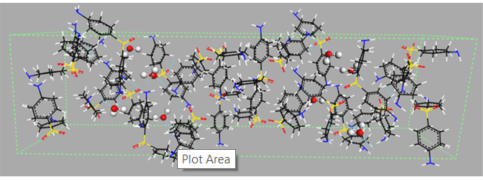
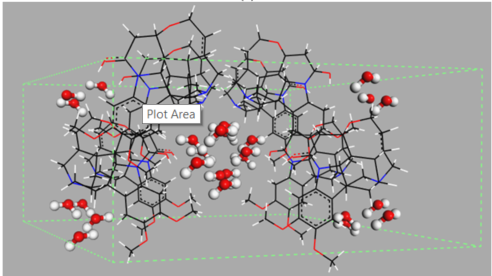
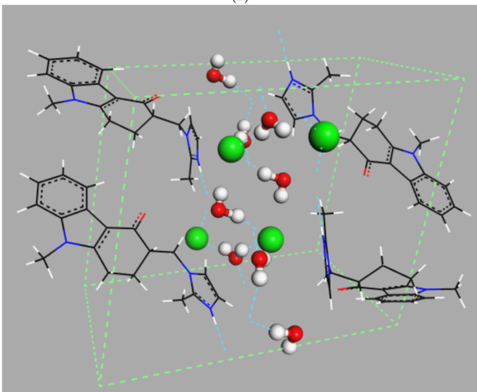
Figure 1. Examples of different types of hydrates: (a) isolated hydrate, dapsone; (b) channel hydrate, brucine [11]; and (c) ion-coordinated hydrate, ondansetron hydrochloride [12]. Crystallographic structures were downloaded from the Cambridge Crystallography Data Centre (CCDC) [13] with the following reference codes: ANSFON (dapsone hydrate), YOYZIX (brucine hydrate), and YILGAB (ondansetron hydrochloride dihydrate). Ondansetron hydrochloride: chlorine is marked in green.
Statistically, approximately one-third of pharmaceutical solids exist in at least two forms, differing in the level of hydration [14]. In most cases, in comparison with anhydrous forms, hydrates are thermodynamically more stable under normal conditions. As a result, they are less prone to dissolve in water and, consequently, they usually exhibit lower bioavailability, which is an obvious disadvantage in terms of their therapeutic applications [15]. This key property can be improved by the formation of hydrate co-crystals [16] or application of selected excipients [17][18]. On the other hand, hydrates show better compressibility and tabletability than anhydrates, are less affected by wet granulation process, and are less susceptible for the tablet storage conditions like temperature and relative humidity (RH). In such cases, it could be reasonable to find and produce the most stable API hydrate to prevent the form alteration during production or storage. The choice between a hydrated and anhydrous form of an API is very often a critical factor for solid dosage forms’ stability and their mechanical behavior [19][20][21].
Nevertheless, there are some exceptions from the above given principles. For example, erythromycin dihydrate shows a more efficient dissolution rate than the respective monohydrate and anhydrate [22][23]. Theophylline monohydrate exhibits better solubility in water than its anhydrous form [24]. These cases indicate the variability in properties of hydrated APIs and necessity to perform comparative studies between the differently hydrated forms in each particular case.
While, from the legal point of view in most countries, for each polymorphic form of an API, a separate patent may be assigned, the situation is more complicated for hydrates. More specifically, in some countries, the given two forms of API differing in hydration level can be perceived as legally the same forms, while this is not the case in others. For example, varying in hydration levels, different forms of cefdinir have received individual patent claims in the United States [25]. By contrast, however, the Brazilian and Argentine Patent Law Guidelines exclude the possibility to patent solvates or hydrates, because they both consider them to be discoveries and not new inventions [26][27]. Nonexistence of a worldwide coherent law on this topic has already brought court trials.
Equally disordered is hydrates’ representation in pharmacopoeias, as some of the hydrates are included in separate monographs and some are not, without any clear justification. In pharmacopoeias, one can find only those hydrated forms of drugs that are most commonly used, as well as hydrates of widely applied excipients like lactose monohydrate [28]. For example, in the International Pharmacopeia [29], one can find separate monographs for both caffeine anhydrate and caffeine monohydrate. However, in the case of carbamazepine, a monograph for the anhydrous form only can be found, even though carbamazepine dihydrate is a well-known and commonly applied form of this API [30].
The above shortly enumerated various issues regarding APIs’ hydrates indicate the complexity of the discussed subject, which can also be a real challenge for the pharmaceutical industry. However, when properly employed, the ability of most of the APIs to form solid hydrates can be also a unique opportunity to improve their stability, processability, or biopharmaceutical properties or even to patent a new solid state form, similarly as in the case of polymorphs. However, for these purposes, a detailed analysis of hydrated pharmaceuticals is essential.
2. Methods Applied for Analysis of Hydrates
2.1. Structure Determination
A starting point in the organic solids analysis is usually detailed determination of their composition and structure. It is especially interesting and important in terms of hydrates, because often, not only polymorphic forms transition, but also change of the hydration degree may occur when the sample is exposed to various temperature, pressure, or humidity conditions.
The difficulty of studies dealing with pharmaceutical hydrates lies in the fact that two different types of molecules are present in a crystal structure, namely API and water. This triggers a stepwise analysis. Firstly, an insight into the number of water molecules per the host substance molecule is needed. This is consistent with contradistinction between stoichiometric and non-stoichiometric hydrates. For this purpose, thermo-analytical method TGA (thermogravimetric analysis) and gravimetric method DVS (dynamic vapour sorption) are usually used. Together with another thermo-analytical method DSC (differential scanning calorimetry), they allow detection of mass gain or loss (due to change of the water content) [31], determination of enantiotropic/monotropic relationship between polymorphs [32], as well as melting or crystallization temperatures [33][34]. Usually, DSC and TGA require a smaller amount of sample (3–10 mg) than DVS (10–30 mg). However, those three methods have some drawbacks and probably the most significant one is the destruction of a sample during the analysis.
It should be also taken into account that these methods not always can be applied for investigation of a mixture of compounds, for example, preformulates or whole drug products in which, besides APIs, excipients are also present. This always depends on a particular example taken into consideration and a type of physicochemical property that aims to be determined.
When a hydration stoichiometry is known, more precise insight into the crystal structure is provided by applying SCXRD (single crystal X ray diffraction), PXRD (powder X ray diffraction), ssNMR (solid state nuclear magnetic resonance spectroscopy), FT-IR (Fourier-transformed infrared spectroscopy), and Raman spectroscopy techniques, as well as computational approaches. They can uniquely identify the crystallinity of API and they are used to determine the exact structure of a hydrated crystal, including the positions and dynamics of water molecules. In the last stage, the analysis of (de)hydration process is sometimes studied. In order to perform it properly, both the knowledge of the structure and results of thermogravimetric approaches are essential.
2.1.1. Structure Determination Techniques
In order to determine the structure of a hydrate, single crystal X-ray diffraction (SCXRD) is usually used. It is the most informative, especially when determining the accurate atoms positions, but at the same time, it is a demanding method. This is because of the usually higher cost of analysis, but more importantly, because of the requirement of a stable crystal of usually a minimum of 0.1 mm in size [35]. Often, this condition cannot be fulfilled, sometimes solely owing to the nature of the sample, as many organic solids do not form stable crystals of the size proper for the SCXRD analysis.
SCXRD is a non-destructive and most willingly used method applied in order to solve a crystal structure. It should be noticed, however, especially in the analysis of hydrates, that the proper determination of the hydrogen atoms’ positions may be very difficult for several reasons. First, the hydrogen atom has only one electron, and thus a very low scattering factor. Further, the electron density distributions from this one electron around hydrogen atoms are usually displaced or pulled towards the bonding regions. Another reason may be the dynamics in the crystal lattice in general and particularly motions involving H atoms that form hydrogen bonds. Finally, the number of water molecules, and thus H atoms forming them, may vary or be significantly disordered in the non-stoichiometric hydrates.
One of the most commonly applied methods in the crystal structure studies is powder X-ray diffraction (PXRD). PXRD delivers data on the unit cell parameters, and thus sometimes serves as an alternative route to a single crystal X-ray diffraction (SCXRD) when the latter is not attainable [36]. Further, in contrast to SCXRD, PXRD can be applied to study the mixture of solids and, in some cases, determine the quantitative phase composition [37]]. More specifically, in terms of hydrate investigation, PXRD helps to differentiate between the hydrated and dehydrated form of the substance. This is plainly visible as a drastic change of a pattern on the diffractogram [38] (Figure 2).
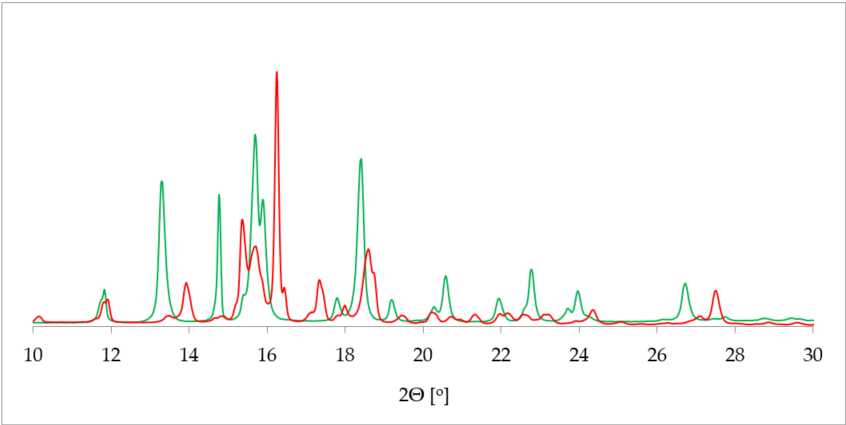
Figure 2. Comparison of powder X-ray diffraction (PXRD) diffractograms of 17-β-estradiol anhydrate (red) received by heating the 17-β-estradiol hemihydrate (green). Source: author’s archive, more details in [38].
However, PXRD requires complicated interpretation and demanding refinement in order to provide such detailed information on the crystal structure as SCXRD, especially in terms of the atoms’ positions in the unit cell. Nevertheless, applying synchronically different techniques, with an exclusion of highly demanding SCXRD, it has already been successfully used to determine the structure of new crystalline compounds, including APIs’ hydrates [39][40][41]. What is more, PXRD patterns can be quickly calculated based on the crystal structure files with a high accuracy using specialized software (Reflex) [42]. Such an approach can be used to refine the experimentally obtained data or to differentiate between various polymorphic forms, including hydrates.
While diffraction-based analytical methods, both PXRD and SCXRD, can be used in the cases of crystalline systems, the next method, solid state NMR, can be used to study both crystalline and amorphous materials (Figure 3).
Solid state NMR (ssNMR) represents a totally different approach than the diffraction methods. It is a non-destructive technique that allows qualitative and in some cases also quantitative measurements. However, ssNMR analysis is relatively expensive and has a long data acquisition time [43]. Intuitively, when applying ssNMR to the study of pharmaceutical hydrates, one could think of registering the 1H or 17O spectra. However, those kinds of experiments are rarely performed because of the broad overlapping signals in the solid state 1H NMR spectra and quadrupolar character of the 17O nucleus. A very promising way to overcome the problem of broad signals in the solid state 1H NMR spectra is ultra-fast magic angle spinning (MAS). For example, this approach has recently been successfully used to differentiate the anhydrous and monohydrate form of theophylline [44]. The most commonly performed ssNMR analysis experiments are 13C and 15N, combined with MAS and cross-polarization (CP). In the cases of organic solids, those approaches can be used to distinguish between the forms at different hydration levels (Figure 3), though such experiments do not provide any direct information on water within the crystal structure. Moreover, especially to study the hydration and dehydration processes, 2H NMR experiments are being performed using D2O in the crystallization process or exposing hygroscopic samples in the contact with the vapor of D2O [45]. However, while this approach can be used to investigate the hydrogen bond dynamics in a crystal lattice, it should be noticed that, because of the isotope effect, both the structure and kinetics of hydration/dehydration may be different when D2O is used instead of H2O.
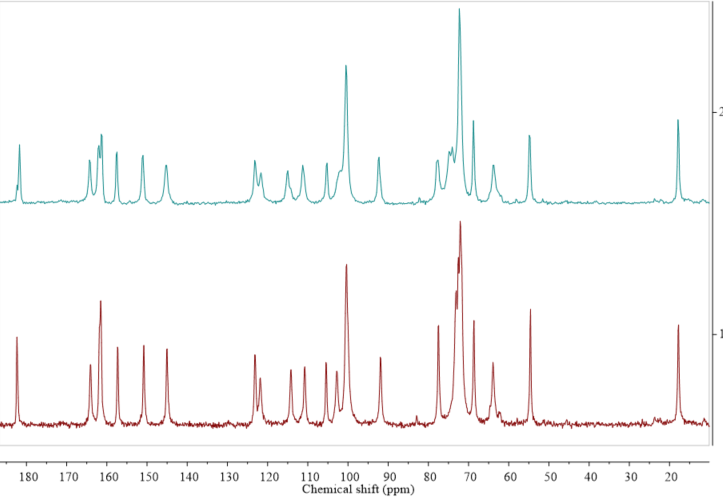
Figure 3. Comparison of 13C cross-polarization (CP) magic angle spinning (MAS) solid state nuclear magnetic resonance spectroscopy (ssNMR) spectra of diosmin monohydrate (red, 1) and anhydrous diosmin (blue, 2). Source: author’s archive, more details in [46].
ssNMR measurements are often combined with precise quantum chemistry calculations, which facilitates or even enables proper assignment of NMR signals [47]. Application of DFT (density functional theory)-based GIAO [48] or GIPAW [49] methods makes it possible to theoretically calculate the NMR parameters, such as chemical shielding constants and anisotropy tensors. Thus, it enables validation or refinement of the suggested crystal structure. The high accuracy of such an approach has been already acknowledged multiple times [50][51][52]. For example, calculations of NMR shielding constants in order to simulate the NMR spectra using available crystal structures enable rapid confirmation of whether the analyzed crystal form of a compound is a hydrate or anhydrous one, without the need for any external standards (Figure 4). Nowadays, such a combination of ssNMR and computational methods is so widely used that such an approach has already been defined as ‘NMR crystallography’ [53].
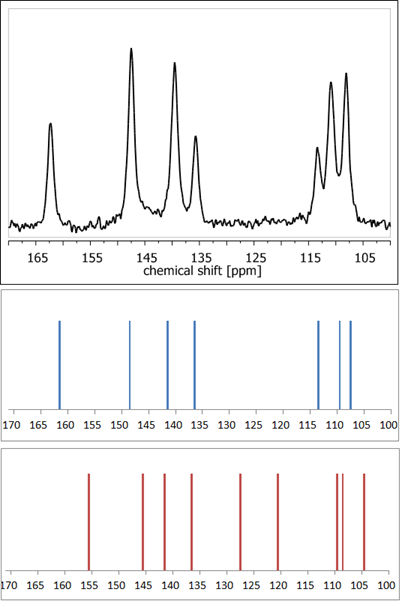
Figure 4. 13C NMR spectra: experimental of anhydrous elagic acid (black) and simulated using CASTEP GIPAW density functional theory (DFT) calculations results for anhydrous elagic acid (blue) and elagic acid dehydrate (red). Source: author’s archive, more details in [54].
Apart from PXRD and NMR described above, probably the quickest and easiest methods used to investigate hydrates are FT-IR and Raman spectroscopies, which are complementary to each other. The presence of the unique bands makes it possible to doubtlessly recognize a crystal substance [55]. These two methods are of a special utility in research performed on hydrates because the presence of water can be recognized owing to signals from the hydroxyl group. The representative stretching band of the hydroxyl group appears at a frequency above 3000 cm-1 [56]. Another interesting aspect is investigation of a hydrogen bonds’ net created as a result of the presence of -OH groups. Often, a relationship between the peak position of such an H-bond and its strength can be defined. Typically, the higher the wavenumber of the O-H peak (even about 3500 cm-1 [57]), the less strong the bond between water and a host molecule in the given hydrate. This, in turn, delivers a suggestion on the overall hydrate structure, because, depending on the character of a host substance, either channel or void hydrate is better stabilized. Consequently, FT-IR and Raman spectroscopies enable analysis of changes in hydrogen bonding that occur during hydration/dehydration process. A good example is rearrangement of this network when estradiol hemihydrate undergoes dehydration (Figure 5) [58]. Moreover, these methods can be applied to identify different polymorphic crystal forms among hydrates. It is based on a careful analysis of the fingerprint spectral region.
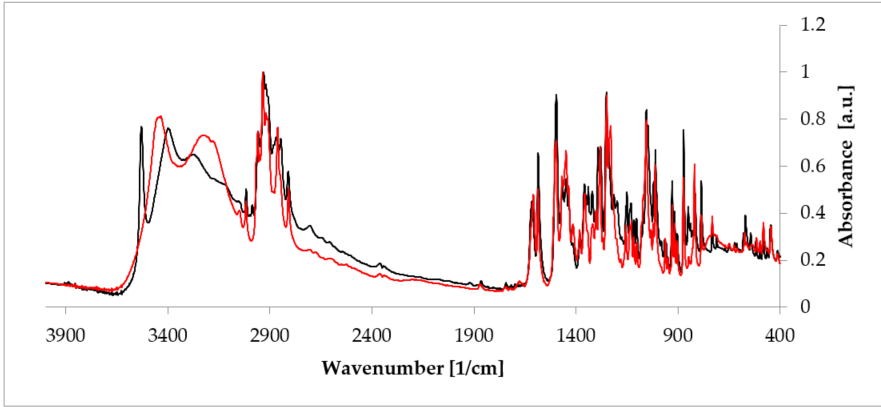
Figure 5. Comparison of the Fourier-transformed infrared spectroscopy (FT-IR) spectra of anhydrous estradiol (black) and estradiol hemihydrate (red). Source: author’s archive, more details in [38].
However, when solely applying FT-IR or Raman spectroscopy, it is not possible to determinate a completely unknown structure. Moreover, especially in the FT-IR transmission technique, because of the longer preparation process, a risk is posed by the environmental humidity, which could alter the analysis [59]. This is why, for the highly absorbing probes, ATR-IR (attenuated total reflectance) is used as it requires no sample preparation and is also a non-destructive method [60].
A combination of the above mentioned techniques helps to differentiate between various types of hydrates, mainly between channel and isolated site hydrates. In case of the first ones, FT-IR shows sharp OH-bands at relatively low frequencies, while in TGA, rather wide weight loss ranges are observed, and DSC presents broad endothermic peaks. On the contrary, ion isolated hydrates are characterized by sharp dehydration endotherms in DSC and narrow weight loss ranges in TGA [61][62].
A completely different approach to determine the structure of small organic molecules is presented by the new method: cryo-electron microscopy/microcrystal electron diffraction CryoEM/MicroED [63]. It enables collecting the high-quality MicroED data from nanocrystals, which results in atomic resolution (<1 Å) crystal structures. CryoEM/MicroED requires a small amount of sample, very simple preparation, and can be conducted in a few minutes. One of its most important advantages is the fact that it can be applied to derive a crystal structure from seemingly amorphous powders.
2.1.2. Purely Computational Structure Determination Techniques
Crystal structure prediction (CSP) is an already widely accepted multi-step methodology used to computationally determine the structure of a crystalline material [64][65][66][67]. In the field of hydrate studies, it can be used to predict the crystal structure of a hydrate of known stoichiometry. At first, molecular mechanics (MM) is used in order to generate and rank possible compound conformations. Afterwards, selected conformers are subjected to quantum mechanics calculations. The former are performed usually on single molecules, and the latter on whole crystal structures. This enables observation of conformational polymorphism [68] in the first case and packing polymorphism [68] in the second. The (calculations performed at temperature 0 K) lattice energies derived in this process could be qualified if kinetics factors (like temperature) were included. For that purpose, time- and computational power-consuming molecular dynamics (MD) must be applied [69]. Including the entropy input is of a high importance because it means that the zero-point energy (ZPE) will be calculated. It has been proven that the ZPE contributes the most to the free energy differences between hypothetical similar structures [70]. Consequently, its neglection leads to the overestimation of the number of structures generated [71]. Furthermore, neglection of the entropy contribution prevents the investigation of the enantiotropically-related polymorphs [72].
Lattice energy is calculated on the basis of the inter- and intra-molecular forces. For example, in the Polymorph Predictor software [73], which is one of the packages used for performing CSP, a two-step procedure is implemented. Firstly, various molecular conformations are generated, and afterwards, optimal hydrogen positions or chemical groups orientations are defined. This process delivers both inter- and intra-molecular energies of the investigated structure, which together form lattice energy.
The target of CSP application in the studies of hydrates is the same as in the case of other molecules; that is, it helps to elucidate the structural information obtained from the experiment, and proposes a variety of structural models that could possibly be the new thermodynamic stable hydrates or polymorphs of the existing hydrates [74].
CSP is usually used when there is no structural information on the investigated molecule available. Its results direct the future experimental approach or support the previous assumptions on the existence and/or structure of the hydrate, as has happened in the case of orotic acid crystal forms [75]. Application of CSP enabled to characterize the structural disorder, and showed stacking faults as well as small variability in packing indices of the anhydrous orotic acid. Another purpose to apply CSP is a situation wherein the experimental attempts to obtain a proper crystal for the SCXRD measurements have failed. In such a case, CSP delivers viable structures that can serve as an important point of reference for further investigations performed with techniques other than SCXRD [76]. A good example is usage of the CSP- and NMR-based approach for analysis of catechin methanol hemisolvate-monohydrate [77]. In this particular study, an important feature of the computational CSP techniques has been utilized. Namely, the possibility to introduce manual changes into the analyzed crystalline systems. This allows to investigate what is not possible regarding hemihydrates in the automatic procedure of calculations [78].
2.2. Kinetics of (De)Hydration Process
The description of a (de)hydration process is mostly based on both experimental and calculation methods. The former encompasses spectroscopic and thermogravimetric methods as well as microscopy and other techniques like Karl–Fisher titration (K–F titration) [79]. This titration enables to determine whether water is adsorbed or incorporated in a crystal structure, the latter meaning that the investigated sample forms a hydrate. It is known in three variants: volumetric, coulometric, and oven coulometric K–F titration [80]. In all cases, the used iodine is proportional to the amount of water, which is calculated on the basis of the titrator consumed during the titration process. To perform oven coulometric K–F titration, the sample is pre-weighted and heated. It is the water vapor that is used in the titration [81]. In this last example, thermogravimetric methods are used in order to investigate the thermal stability of a probe and determine the minimal temperature needed for the oven coulometric K–F approach. In general, K–F titration is a very sensitive method. It is found in the pharmacopeial monographs of hydrates, for example, terpin hydrate [82].
If, during the examination of the process with the application of the thermogravimetric methods, big hysteresis in dehydration is observed, it may suggest that the obtained anhydrate is a stable one and it can be analyzed in detail separately by TGA, PXRD, FT-IR, and so on. However, when dehydration is defined as reversible, application of combination of different methods, including Karl–Fischer titration, is recommended.
For proper (de)hydration research, an insight into H-bonding structure is needed. Apart from the already mentioned ssNMR and ‘NMR crystallography’, purely computational approaches are also undertaken. For that purpose, DFT calculations involving application of hybrid functionals (most commonly: B3LYP, M05) and polarizable function (an extended basis set like 6-31++G(d,p) or 6-311+G(2d,2p)) are performed [83][84][85][86][87]. Especially, the latter plays a crucial role, as polarizable functions are used to properly define the H-bonding. This helps not only to calculate the energy of the intermolecular interactions (performed also in CSP approach), but also, in further steps, to visualize the H-bonding pattern. Often, it facilitates explanation of the observed (de)hydration mechanism.
In a computational investigation of the (de)hydration kinetics, an important role is also played by calculation of the solvent-accessible volume, which significantly differs for non-stoichiometric and stoichiometric hydrates [88]. Resulting from water loss, possible collapse of a void or conformational disorientation has a direct impact on the above discussed H-bonds network.
2.3. Stability Determination in the Industrial Production
The analysis of (de)hydration kinetics is performed to describe the reaction process and to determine whether a transformation between the differently hydrated forms under the given conditions would take place spontaneously, and if so, at what rate [89][90]. The decisive parameters are the energy barrier of water diffusion into the lattice as well as the energy of conformational change. Conversely, the thermodynamic stability investigation is based solely on the reactants of the given process. Described as a function of the Gibbs energy change (Gibbs enthalpy, ΔG) [91], for pharmaceutical purposes, thermodynamic stability is calculated in isothermal-isobaric conditions (NPT ensemble) [92]. Along with molecular dynamics (MD) or compound-water binding energy calculations [93], it can determine the temperature and pressure values under which the global minimum could be achieved. For the pharmaceutical industry, these data are of crucial importance and are equivalent with occurrence of the polymorphic phase transition or desolvation, which could appear during product manufacturing or its storage. These calculations enable to establish a stability order among very often numerous (de)hydrated and amorphous forms of API.
Often, calculations are performed with software based on DFT-D (dispersion correction). Such an approach is already known for high accuracy [94]. However, it must be emphasized that computing the energy difference between hydrate and anhydrate is more challenging than computation of the already relatively well-described polymorph transitions [95]. The reason is that, most commonly applied and so far probably most accurate for this purpose, PBE functional overestimates the energy of a transition from hydrate to anhydrate [96].
In order to experimentally determine the substance stability, measurement of relative humidity (RH) and water activity aw is applied. As aw informs about the substance solubility [97], its value has a connection with hydrates’ stability order. According to the ICH Guidance for Industry: Q1A(R2) Stability Testing of New Drug Substances and Products, stability measurements for non-sterile products during long-term storage should be performed at specified conditions, which are as follows: 25 °C/60% RH and aw < 0.60. This is because RH = 60% and aw = 0.60 are established as border values for microorganisms’ growth [98]. Regular and accurate testing in the topic is of great importance as only a proper adjustment of RH, aw and temperature parameters allows control of the phase formation process and, as a consequence, delivers a stable product [99].
References
- Brog, J.-P.; Chanez, C.-L.; Crochet, A.; Fromm, K.M. Polymorphism, what it is and how to identify it: A systematic review. RSC Advances 2013, 3, 16905–16931.
- Tieger, E.; Kiss, V.; Pokol, G.; Finta, Z.; Dušek, M.; Rohlíček, J.; Skořepová, E.; Brázda, P. Studies on the crystal structure and arrangement of water in sitagliptin L-tartrate hydrates. CrystEngComm 2016, 18, 3819–3831.
- Braun, D.E.; Gelbrich, T.; Kahlenberg, V.; Laus, G.; Weiser, J.; Griesser, U.J. Packing polymorphism of a conformationally flexible molecule (aprepitant). New J. Chem. 2008, 32, 1677–1685.
- Seddon, K.R. Pseudopolymorph: A Polemic. Cryst. Growth Des. 2004, 4(6), 1087.
- Bernstein, J. Another Comment on Pseudopolymorphism. Cryst. Growth Des. 2005, 5, 1661–1662.
- Tian, F.; Qu, H.; Louhi-Kultanen, M.; Rantanen, J. Insight into Crystallization Mechanisms of Polymorphic Hydrate Systems. Chem Engineering and Tech 2010, 33, 833–838
- Fucke, K.; Steed, J.W. X-ray and Neutron Diffraction in the Study of Organic Crystalline Hydrates, Water, 2010, 2, 333–350.
- Authelin, J.-R. Thermodynamics of non-stoichiometric pharmaceutical hydrates. Int. J. of Pharm. 2005, 303, 37–53.
- Gossman, W.L.; Wilson, S.R.; Oldfield, E. Three hydrates of the bisphosphonate risedronate, consisting of one molecular and two ionic structures. Acta Crystallogr. Sect. C Cryst. Struct. Commun. 2003, 59, m33–m36, doi:10.1107/s0108270102021996.
- Zimmermann, A.; Tian, F.; De Diego, H.L.; Frydenvang, K.; Bar-Shalom, D.; Elema, M.R.; Hovgaard, L. Structural Characterisation and Dehydration Behaviour of Siramesine Hydrochloride. J. Pharm. Sci. 2009, 98, 3596–3607, doi:10.1002/jps.21679.
- Braun, D.E; Griesser, U.J. Stoichiometric and Nonstoichiometric Hydrates of Brucine. Cryst. Growth Des. 2016, 16, 6111–6121
- Collin, S.; Moureau, F.; Quintero, F.M.; Vercauteren, D.P.; Evrard, G.; Durant, F. Stereoelectronic requirements of benzamide 5HT3 antagonists. Comparison with D2 antidopaminergic analogues. J. Chem. Soc. Perkin Trans. 1995, 2, 77.
- Cambridge Crystallographic Data Centre. Available online: https://www.ccdc.cam.ac.uk/, (accessed on 01 September 2020).
- Ma, X.; Müller, F.; Huang; S.; Lowinger, M.; Liu, X.; Schooler, R.; Williams, R.O.III. Influence of carbamazepine dihydrate on the preparation of amorphous solid dispersions by hot melt extrusion. Pharmaceutics. 2020, 12, 379.
- Censi, R.; Di Martino, P. Polymorph Impact on the Bioavailability and Stability of Poorly Soluble Drugs. Molecules. 2015, 20, 18759–18776.
- Sathisaran, I.; Dalvi, S.V. Engineering Cocrystals of Poorly Water-Soluble Drugs to Enhance Dissolution in Aqueous Medium. Pharmaceutics. 2018, 10, 108.
- Tian, F.; Qu, H.; Zimmermann, A.; Munk, T.; Jørgensen, A.C.; Rantanen, J. Factors affecting crystallization of hydrates. J. of Pharm. Pharmacol. 2010, 62, 1534–1546
- Scaramuzza, D.; Shneider Rauber, G.; Voinovich, D.; Hasa, D. Dehydration without Heating: Use of Polymer-Assisted Grinding for Understanding the Stability of Hydrates in the Presence of Polymeric Excipients. Cryst. Growth Des. 2018, 18, 5245–5253.
- Brittain, G.H. Polymorphism and Solvatomorphism. J. of Pharm. Sc. 2010, 99, 3648–3664
- Giron, D.A.; Mutz, M.; Goldbronn, C.; Pfeffer, S.; Piechon. P.; Schwab, P. Solid State Characterizations of Pharmaceutical Hydrates. J. of Therm. Anal. Calorim. 2002, 68, 453–465
- Sood, J.; Sapra, B.; Bhandari, S.; Jindal, M.; Tiwary, A.K. Understanding pharmaceutical polymorphic transformations I: Influence of process variables and storage conditions. Ther.Del. 2014, 5, 1123–1142.
- Allen, P.V.; Rahn, P.D.; Sarapu, A.C.; Vanderwielen, A.J. Physical characterization of erythromycin: anhydrate, monohydrate, and dihydrate crystalline solids. J Pharm Sci. 1978, 67, 1087–1093.
- Blagden, N.; de Matas, M.; Gavan, P.T.; York, P. Crystal engineering of active pharmaceutical ingredients to improve solubility and dissolution rates. Adv. Drug Deliv. Rev. 2007, 59, 617-630.
- Bobrovs, L.; Seton, L.; Dempster, N. The reluctant polymorph: investigation into the effect of self-association on the solvent mediated phase transformation and nucleation of theophylline. CrystEngComm. 2015, 17, 5237-5251
- Law, D.; Henry, R.; Lou; X. Trihemihydrate, anhydrate and novel hydrate forms of Cefdinir. U.S. Patent Application No. 11/072,568, US20060025399A1, 01 September 2020.
- Brazil’s Patent Reform Innovation Towards National Competitiveness, p. 124. Avaliable online: http://infojustice.org/wp-content/uploads/2013/09/Brazilian_Patent_Reform.pdf (accessed on 1 September 2020).
- Ho, C.M. Should All Drugs Be Patentable?: A Comparative Perspective. Vand. J. Ent. & Tech. L. 2014, 17, 295–348.
- Lactose Monohydrate Monographia in the US Pharmacopoeia. Available online: https://www.usp.org/harmonization-standards/pdg/excipients/lactose-monohydrate (accessed on 1 September 2020).
- Caffeine anhydrous, Caffeine monohydrate monographs in the International Pharmacopoeia 9th Edition. Available online: https://apps.who.int/phint/en/p/docf/ (accessed on 01 September 2020).
- Flicker, F.; Eberle, V.A.; Betz, G. Variability in commercial carbamazepine samples – Impact on drug release. Int. J. of Pharm. 2011, 410, 99–106.
- Forster, A.; Gordon, K.; Schmierer, D.; Soper, N.; Wu, V.; Rades, T. Characterisation of two polymorphic forms of ranitidine-HCl, Internet J. Vib. Spectrosc. 2 1998, 2, 12.
- Burger, A.; Ramberger, R. On the polymorphism of pharmaceuticals and other molecular crystals. II. Microchim. Acta, 1979, 72, 273–316.
- Slobodin, B.V.; Samigullina, R.F. Thermoanalytical study of the polymorphism and melting behavior of Cu2V2O7. Inorg. Mat. 2010, 46, 196–200.
- Han, J.; Suryanarayanan, R. A method for the rapid evaluation of the physical stability of pharmaceutical hydrates. Thermochim. Acta 1999, 329, 163–170.
- Prendergast, R. Structure determination of small and large molecules using single crystal X-ray crystallography. Master’s Thesis, University of Manchester, Manchester, UK, 2010.
- Bukovec, P.; Meden, A.; Smrkolj, M.; Vrecer, F. Influence of Crystal Habit on the Dissolution of Simvastatin Single Crystals. Acta Chim. Slov. 2015, 62, 958–966.
- Kirchmeyer, W.; Grassmann, O.; Wyttenbach, N.; Alsenz, J.; Kuentz, M. Miniaturized X-ray Powder Diffraction Assay (MixRay) for Quantitative Kinetic Analysis of Solvent-Mediated Phase Transformations in Pharmaceutics. J. Pharm. Biomed. Anal. 2016, 131, 195–201.
- Szeleszczuk, Ł.; Jurczak, E.; Zielińska-Pisklak, M.; Harwacki, J.; Pisklak, D.M. Comparison of the analytical methods (solid state NMR, FT-IR, PXRD) in the analysis of the solid drug forms with low concentration of an active ingredient - 17-β-estradiol case. J. Pharm. Biomed. Anal. 2018, 149, 160–165.
- Nugrahani, I.; Slamet, I.; Mauludin, R.; Almira, M. Hydrate transformation study of fluoroquinolone antibiotics using Fourier transform infrared spectroscopy (FTIR). Int. J. of Pharm. Pharm. Sc. 2015, 7, 246–252.
- Lu, J.; Wang, J.; Li, Z.; Rohani, S. Characterization and pseudopolymorphism of Lphenylalanine anhydrous and monohydrate forms. Afr. J. of Pharm. Pharmacol. 2012, 6, 269–277.
- Seton, L.; Khamar, D.; Bradshaw, I.J.; Hutcheon, G.A. Solid State Forms of Theophylline: Presenting a New Anhydrous Polymorph. Cryst. Growth Des. 2010, 10, 3879–3886.
- MS Reflex Plus module within the Accelrys Materials Studio. Available online: https://www.3ds.com/fileadmin/PRODUCTS-SERVICES/BIOVIA/PDF/BIOVIA-material-studio-reflex-plus.pdf (accessed on 1 September 2020).
- Tishmack,P.A.; Bugay, D.A.; Byrn, S.R. Solid-state nuclear magnetic resonance spectroscopy – pharmaceutical application, J. Pharm. Sci. 2003, 92, 441–474.
- Hirsh, D.A.; Wijesekara, A.V.; Carnahan, S.L.; Hung, I.; Lubach, J.W.; Nagapudi, K.; Rossini, A.J. Rapid Characterization of Formulated Pharmaceuticals Using Fast MAS 1H Solid-State NMR Spectroscopy. Mol. Pharm. 2019, 16, 3121–3132.
- Chan-Huot, M.; Wimperis, S.; Gervais, C.; Bodenhausen, G.; Duma, L. Deuterium MAS NMR Studies of Dynamics on Multiple Timescales: Histidine and Oxalic Acid, Chem. Phys. Chem. 2014, 16, 204–215.
- Szeleszczuk, Ł.; Pisklak, M.; Zielińska-Pisklak, M.; Wawer, I. Spectroscopic and structural studies of the diosmin monohydrate and anhydrous diosmin. Int. J. of Pharm. 2017, 529, 193–199.
- Gowda, V.; Laitinen, R.S.; Telkki, V.-V.; Larsson, A.C.; Antzutkin, O.N.; Lantto, P. DFT calculations in the assignment of solid-state NMR and crystal structure elucidation of a lanthanum(III) complex with dithiocarbamate and phenanthroline. Dalton Trans. 2016, 45, 19473–19484.
- Costa, F.L.P.; Gomez, P.F.; Silva, K.A.; Liao, L.M. Conformational Analysis, Experimental and GIAO-DFT 13C NMR Chemical Shift Calculation on 2′-Hydroxy-3,4,5-trimethoxy-chalcone. J. Braz. Chem. Soc. 2017, 28, 2130–2135.
- Charpentier, T. The PAW/GIPAW approach for computing NMR parameters: A new dimension added to NMR study of solids. Solid State NMR 2011, 40(1), 1–20.
- Iron, M.A. Evaluation of the Factors Impacting the Accuracy of 13C NMR Chemical Shift Predictions using Density Functional Theory—The Advantage of Long-Range Corrected Functionals. J. Chem. Theory Comput. 2017, 13, 5798–5819
- Gao, P.; Zhang, J.; Peng, Q. Glezakou, A.-V. A General Protocol for the Accurate Predictions of Molecular 13C/1H NMR Chemical Shifts via Machine Learning. ChemRxiv 2019.
- Czernek, J.; Brus, J. Exploring Accuracy Limits of Predictions of the 1H NMR Chemical Shielding Anisotropy in the Solid State. Molecules. 2019, 24, 1731.
- Bryce, D.L. NMR crystallography: structure and properties of materials from solid-state nuclear magnetic resonance observables. IUCrJ 2017, 4, 350–359.
- Szeleszczuk, Ł.; Pisklak, D.M.; Zielińska-Pisklak, M.; Wawer, I. Effects of structural differences on the NMR chemical shifts in cinnamic acid derivatives: Comparison of GIAO and GIPAW calculations. Chem. Phys. Lett. 2016, 653, 35-41.
- Colthup, N.L.; Daly, L.H.; Wiberley, S.E. Introduction to Infrared and Raman Spectroscopy, 3rd Ed.; Academic Press, Inc.: San Diego, USA, 1990.
- Biswas, R.; Carpenter, W.; Voth, G.A.; Tokmakoff, A. Molecular modeling and assignment of IR spectra of the hydrated excess proton in isotopically dilute water. J. Chem. Phys. 2016, 145, 154504.
- Hansen, P.E.; Spanget-Larsen, J. NMR and IR Investigations of Strong Intramolecular Hydrogen Bonds. Molecules 2017, 22, 552.
- Van Eerdenbrugha, B.; Taylor, L.S. Application of mid-IR spectroscopy for the characterization of pharmaceutical systems. Int. J. of Pharm. 2010, 417, 3–16
- Szakonyi, G.; Zelko, R. The effect of water on the solid state characteristics of pharmaceutical excipients: Molecular mechanisms, measurement techniques, and quality aspects of final dosage form. Int. J. Pharm. Investig. 2012, 2, 18–25.
- Oliver, K.V.; Marechal, A.; Rich, P.R. Effects of the Hydration State on the Mid-Infrared Spectra of Urea and Creatinine in Relation to Urine Analyses. Appl. Spectrosc. 2016, 70, 983–994.
- Healy, A.M.; Worku, Z.A.; Kumar, D.; Madi, A.M. Pharmaceutical solvates, hydrates and amorphous forms: A special emphasis on cocrystals. Adv. Drug Deliver. Rev. 2017, 117, 25–46.
- Feth, M.P.; Nagel, N.; Baumgartner, B.; Bröckelmann, M.; Rigal, D.D.; Otto, B.; Spitzenberg, M.; Schulz, M.; Becker, B.D.; Fischer, F.; Petzoldt, C. Challenges in the development of hydrate phases as active pharmaceutical ingredients--an example. Eur. J. Pharm. Sci. 2011, 42, 116–129.
- Jones, C.G.; Martynowycz, M.W.; Hattne, J.; Fulton, T.J.; Stoltz, B.J.; Rodriguez, J.A.; Nelson, H.M.; Gonen, T. The CryoEM Method MicroED as a Powerful Tool for Small Molecule Structure Determination. ACS Cent Sci. 2018, 4, 1587–1592.
- Vasileiadis, M.; Pantelides, C.C.; Adjiman, C.S. Prediction of the crystal structures of axitinib, a polymorphic pharmaceutical molecule. Chem. Eng. Sci. 2015, 121, 60–76.
- Baias, M; Dumez, J. N.; Svensson, P. H.; Schantz, S.; Day, G. M.; Emsley, L. De Novo Determination of the Crystal Structure of a Large Drug Molecule by Crystal Structure Prediction-Based Powder NMR Crystallography. J. Am. Chem. Soc. 2013, 135, 17501–17507.
- Woodley, S.M. Crystal structure prediction from first principles. Nat. Mater. 2008, 7, 937–946.
- Hoja, J.; Ko, H-Y.; Neumann, M.A.; Car, R.; DiStasio, R.A.; Tkatchenko, A. Reliable and practical computational description of molecular crystal polymorphs. Sci. Adv. 2019, 5, eaau3338.
- Ratkova, E.L.; Abramov, Y.; Baskin, I.L.; Livingstone, D.J.; Fedorov, M.V.; Withnall, M.; Tetko, I.V. Background Information on Physical -Chemical Properties- Polymorphism. In Comprehensive Medicinal Chemistry III. 3rd ed.; Ed. Chackalamannil, S., Rotella, D., Ward, S.E., Eds.; Elsevier: Oxford, UK, 2017, pp. 396–400.
- Durrant, J.D.; McCammon, J.A. Molecular dynamics simulations and drug discovery. BMC Biol. 2011, 9, 71.
- Abramov, Y.A. Current Computational Approaches to Support Pharmaceutical Solid Form Selection. Org. Process Res. Dev. 2013, 17, 472−485.
- Day, G.M. Current approaches to predicting molecular organic crystal structures. Crystal. Rev. 2011, 17, 3–52.
- Nyman, J.; Graeme, M.D. Modelling temperature-dependent properties of polymorphic organic molecular crystals. Phys. Chem. Chem. Phys. 2016, 18, 31132–31143.
- Polymorph Predictor BIOVIA Materials Studio. Available online: https://www.3ds.com/fileadmin/PRODUCTS-SERVICES/BIOVIA/PDF/BIOVIA-material-studio-polymorph-predictor.pdf, (accessed 01 September 2020).
- Braun, D.E.; Nartowski, K.P.; Khimyak, Y.Z.; Morris, K.R.; Byrn, S.R.; Griesser, U.J.. Structural Properties, Order−Disorder Phenomena, and Phase Stability of Orotic Acid Crystal Forms. Mol. Pharm. 2016, 13, 1012–1029.
- Dudek, M.K. Paluch, P., Pindelska, E. Crystal structures of two furazidin polymorphs revealed by a joint effort of crystal structure prediction and NMR crystallography. Acta Crystall. B 2020, 76, 322–335.
- Dudek, M.K.; Paluch, P.; Śniechowska, J.; Nartowski, K.P.; Day, G.M.; Potrzebowski, M.J. Crystal structure determination of an elusive methanol solvate – hydrate of catechin using crystal structure prediction and NMR crystallography. CrystEngComm 2020, 22, 4969–4981.
- Braun, D.E.; Gelbrich, T.; Griesser, U.J. Experimental and computational approaches to produce and characterise isostructural solvates. CrystEngComm 2019, 21, 5533-5545
- May, J.C.; Grim, E.; Wheeler, R.M.; West, J. Determination of residual moisture in freeze-dried viral vaccines: Karl Fischer, gravimetric and thermogravimetric methodologies. J. Biol. Stand. 1982, 10, 249–259.
- Bruttel, P.; Schlink, R. Water Determination by Karl Fischer Titration, In Pharmaceutical Sciences Encyclopedia; Gad, S.C., Ed. doi:10.1002/9780470571224.pse415
- Kestens, V.; Conneely, P.; Bernreuther, A. Vaporisation coulometric Karl Fischer titration: A perfect tool for water content determination of difficult matrix reference materials. Food Chem. 2008, 106, 1454–1459.
- U.S. Pharmacopoeia, Terpin hydrate monograph. Available online: http://www.pharmacopeia.cn/v29240/usp29nf24s0_m80920.html (accessed: on 25 September 2020)
- Rezende. C.A.; San Gil, R.A.S.; Borré, L.B.; Pires, J.R.; Vaiss, V.S.; Resende, J.A.L.C.; Leitão, A.A.; De Alencastro;Katia, R.B.; Leal, K.Z. Combining Nuclear Magnetic Resonance Spectroscopy and Density Functional Theory Calculations to Characterize Carvedilol Polymorphs. J. of Pharm. Sc. 2015, 105, 2648–2655.
- Hédoux, A.; Paccou,L.; Derollez, P.; Guinet, Y. Dehydration mechanism of caffeine hydrate and structural description of driven metastable anhydrates analyzed by micro Raman spectroscopy. Int. J. Pharm. 2015, 486, 331–338.
- Basford, P.A.; Back, K.R.; Cram, M.; Docherty, R.; Davey, R.J.; Cruz-Cabeza, A.J. Impact of Crystal Structure and Molecular Conformation on the Hydration Kinetics of Fluconazole. Cryst. Growth 2019, 19, 7193–7205.
- Braun, D.E.; Oberacher, H.; Arnhard, K.; Orlova, M.; Griesser, U.J. 4-Aminoquinaldine monohydrate polymorphism: prediction and impurity aided discovery of a difficult to access stable form. CrystEngComm 2016, 22, 4053–4067.
- Kons, A.; Rutkovska, L.; Bērziņš, A.; Bobrovs, R.; Actiņš, A. Three anhydrous forms and a dihydrate form of quifenadine hydrochloride: a structural study of the thermodynamic stability and dehydration mechanism. Cryst EngComm 2015, 17, 3627–3635.
- Braun, D.E.; Griesser, U.J. Why Do Hydrates (Solvates) Form in Small Neutral Organic Molecules? Exploring the Crystal Form Landscapes of the Alkaloids Brucine and Strychnine. Cryst. Growth 2016, 16, 6405–6418.
- Leane, M.M.; Gamble, J.F.; Brown, J.; Hughes, H.; Crull, G.; Engstrom, J.; Gao, Q.; Bunker, M.; Rutherford, S.; Parker, A.; et al. Imaging Dehydration Kinetics of a Channel Hydrate Form of the HIV-1 Attachment Inhibitor Prodrug BMS-663068. J. of Pharm. Sc. 2013, 102, 4375–4383.
- Feth, M. P.; Jurascheck, J.; Spitzenberg, M.; Dillenz, J.; Bertele, G.; Stark, H. New technology for the investigation of water vapor sorption–induced crystallographic form transformations of chemical compounds: a water vapor sorption gravimetry–dispersive raman spectroscopy coupling. J. Pharm. Sci. 2011, 100(3), 1080-1092.
- Goldstein, S.; Lebowitz, J.L.; Tumulka, R.; Zanghi, N. Gibbs and Boltzmann Entropy in Classical and Quantum Mechanics. In Statistical Mechanics and Scientific Explanation: Determinism, Indeterminism and Laws of Nature; Allori, V., Ed.; World Scientific: Singapore, 2020.
- Braun, E.; Gilmer, J.; Mayes, H.B.; Mobley, D.L.; Monroe, J.I.; Prasad, S.; Zuckerman, D.M. Best Practices for Foundations in Molecular Simulations. Living J. Comp. Mol. Sci. 2019, 1, 5957.
- Coates, R.A.; Armentrout, P.B. Binding energies of hydrated cobalt(II) by collision-induced dissociation and theoretical studies: evidence for a new critical size. Phys. Chem. Chem. Phys. 2018, 20, 802–818.
- Wen, S.; Beran, G.J.O. Crystal Polymorphism in Oxalyl Dihydrazide: Is Empirical DFT-D Accurate Enough? J. Chem. Theory Comput. 2012, 8, 2698–2705.
- Rychkov, D.A.; Stare, J.; Boldyreva, E.V. Pressure-driven phase transition mechanisms revealed by quantum chemistry: L-serine polymorphs. Phys. Chem. Chem. Phys. 2017, 19, 6671–6676.
- Thierfelder, C.; Hermann, A.; Schwerdtfeger, P.; Schmidt, W.G. Strongly bonded water monomers on the ice Ih basal plane: Density-functional calculations. Phys. Rev. B 2006, 74, 045422.
- Skowronsky, L.. Water Activity Applications in the Pharmaceutical Industry Ed.1 Cundall, A.; Ed.2 Fontana, A.Jr.Pub. by PDA/DHI, USA, 2009,. 223-252.
- Skovronsky, L. Inhibition of microbial growth in solid dosages at ICH stability storage conditions. Available online: https://www.europeanpharmaceuticalreview.com/article/8876/inhibition-of-microbial-growth-in-solid-dosages-at-ich-stability-storage-conditions/, (accessed on 01 September 2020).
- Touil, A.; Peczalski, R.; Zagrouba, F. Monitoring of theophylline dehydration in a vacuum contact dryer by near-infrared spectroscopy. Chem. Eng. Res. Des. 2013, 91, 1063–1070.
- Chennuru, R.; Koya, R.T.; Kommavarapu, P.; Narasayya, S.V.; Muthudoss, P.; Vishweshwar, P.; Chandra, R.R.; Mahapatra, B.S. In Situ Metastable Form: A Route for the Generation of Hydrate and Anhydrous Forms of Ceritinib. Cryst. Growth Des. 2017, 17, 6341–6352.


