Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Lucas Fornari Laurindo | -- | 3547 | 2022-08-15 12:03:38 | | | |
| 2 | Beatrix Zheng | -48 word(s) | 3499 | 2022-08-16 03:29:53 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Carpi, R.Z.; Barbalho, S.M.; Sloan, K.P.; Laurindo, L.F.; Gonzaga, H.F.; Grippa, P.C.; Zutin, T.L.M.; Girio, R.J.S.; Repetti, C.S.F.; Detregiachi, C.R.P.; et al. Microbiome, Non-Alcoholic Fat Liver Disease and Non-Alcoholic Steatohepatitis. Encyclopedia. Available online: https://encyclopedia.pub/entry/26146 (accessed on 23 July 2026).
Carpi RZ, Barbalho SM, Sloan KP, Laurindo LF, Gonzaga HF, Grippa PC, et al. Microbiome, Non-Alcoholic Fat Liver Disease and Non-Alcoholic Steatohepatitis. Encyclopedia. Available at: https://encyclopedia.pub/entry/26146. Accessed July 23, 2026.
Carpi, Rodrigo Zamignan, Sandra M. Barbalho, Katia Portero Sloan, Lucas Fornari Laurindo, Heron Fernando Gonzaga, Paulo Cesar Grippa, Tereza L. Menegucci Zutin, Raul J. S. Girio, Cláudia Sampaio Fonseca Repetti, Cláudia Rucco Penteado Detregiachi, et al. "Microbiome, Non-Alcoholic Fat Liver Disease and Non-Alcoholic Steatohepatitis" Encyclopedia, https://encyclopedia.pub/entry/26146 (accessed July 23, 2026).
Carpi, R.Z., Barbalho, S.M., Sloan, K.P., Laurindo, L.F., Gonzaga, H.F., Grippa, P.C., Zutin, T.L.M., Girio, R.J.S., Repetti, C.S.F., Detregiachi, C.R.P., Bueno, P.C.S., Pereira, E.D.S.B.M., Goulart, R.D.A., & Haber, J.F.D.S. (2022, August 15). Microbiome, Non-Alcoholic Fat Liver Disease and Non-Alcoholic Steatohepatitis. In Encyclopedia. https://encyclopedia.pub/entry/26146
Carpi, Rodrigo Zamignan, et al. "Microbiome, Non-Alcoholic Fat Liver Disease and Non-Alcoholic Steatohepatitis." Encyclopedia. Web. 15 August, 2022.
Copy Citation
Modifications in the microbiota caused by environmental and genetic reasons can unbalance the intestinal homeostasis, deregulating the host’s metabolism and immune system, intensifying the risk factors for the development and aggravation of non-alcoholic fat liver disease (NAFLD). The use of probiotics, prebiotics and synbiotics have been considered a potential and promising strategy to regulate the gut microbiota and produce beneficial effects in patients with liver conditions.
non-alcoholic fatty liver disease (NAFLD)
non-alcoholic steatohepatitis (NASH)
prebiotic
probiotic
symbiotic
microbiota
1. Introduction
The accelerated industrialization and urbanization processes have evidenced the exponential increase in ultra-processed food consumption and the predominance of a sedentary lifestyle. This setting has developed an epidemic of obesity, dyslipidemia, type 2 diabetes mellitus (DM2), and Metabolic Syndrome (MS) that are associated with the development of non-alcoholic fatty liver disease (NAFLD) that is the most prevalent chronic liver disease worldwide [1][2][3]. The global occurrence of NAFLD is 25.24%, and the highest prevalence rate of the disease is found in the Middle East (31.79%), followed by South America (30.45%), Asia (27.37%), North America (24.13%), Europe (23.71%), and Africa (13.48%) [1][4].
NAFLD is a term that encompasses a cluster of disorders related to the macrovesicular accumulation of triglycerides within hepatocytes higher than 5%, thus causing steatosis. This process occurs due to a mismatch of regulatory mechanisms involved in lipid metabolism. Some patients develop a more aggressive subtype of the disease characterized by lobular inflammation plus hepatic balloon degeneration, with or without fibrosis, known as non-alcoholic steatohepatitis (NASH) [5][6][7].
Approximately 22.5% of NASH patients will develop hepatocellular carcinoma, and 20% will develop cirrhosis [8][9][10][11]. This progressive feature makes NASH the second most prevalent cause of liver transplantation in the USA [12]. Besides, NAFLD has an intense correlation to cardiovascular disease (CVD). A meta-analysis conducted by Haddad et al. showed that the prevalence of cardiovascular events in patients with NAFLD (14.9%) was more than two-fold compared to patients without NAFLD (6.2%) [13].
The burden of NAFLD is increasing worldwide, and its complications are severe. Numerous therapies have emerged to treat or slow the disease’s advance. Some studies have recently shown favourable results with interventions carried out in the intestine that reflect improvements in the liver, such as the bio modulation of intestinal microbiota [14][15][16][17][18]. The use of probiotics, prebiotics and synbiotics has been considered a potential and promising strategy to regulate the gut microbiota [19][20]. The intestinal microbiota is an intense and dynamic ambient whose composition continually changes. These alterations, caused by environmental and genetic reasons, can unbalance the intestinal homeostasis, deregulating the host’s metabolism and immune system, intensifying the risk factors for the development and aggravation of NAFLD [15][21].
Some reviews investigated the effects of the use of prebiotics and probiotics in NAFLD patients, but most evaluated the effectiveness only on hepatic enzymes [22][23]. Other reviews evaluated the effectiveness of prebiotics or probiotics alone [24][25][26][27]. Only two reviews compared the effects of probiotics, prebiotics and synbiotics on liver enzymes, but not on other risk factors associated with NAFDL and NASH [23][28]. Souza et al. [28] also performed a review comparing the effects of probiotics, prebiotics and synbiotics on NAFLD. Nevertheless, only four trials were included in the review.
2. NAFLD and NASH
NAFLD represents a clinicopathological spectrum of liver diseases extending from isolated steatosis fibrosis to cirrhosis and related to hepatocellular carcinoma development. It is defined as the development of steatosis in more than 5% of hepatocytes identified either histologically or radiologically, in the absence of secondary causes such as significant alcohol consumption (<30 g/day for man and <20 g/day for woman), hereditary liver diseases, or viral hepatitis [29]. This disease is comprised of two main entities: NAFLD and NASH. Histologically, the first includes any case characterized by steatosis with minimal or absent lobular inflammation. The second constitutes a more progressive form, characterized by balloon hepatocyte degeneration and diffuse lobular inflammation with or without fibrosis [5]. An increased risk of cirrhosis, hepatocellular carcinoma, and liver-related mortality is associated with NASH, especially when fibrosis is already present. In advanced stages of fibrosis, the mortality rate increases exponentially [30][31].
Although NASH may be suspected in the case of fatty liver and elevated liver enzymes, liver biopsy with histological examination is the only diagnostic method. On the other hand, the diagnosis of NAFLD may be made either by histological examination or by imaging studies that can detect more than 5% of hepatic steatosis. In this sense, ultrasound (US) and computed tomography (CT) are capable of detecting steatosis involving 20% of hepatocytes, and magnetic resonance imaging (MRI) is capable of detecting stenosis in 5% [32][33].
The NAFLD is currently recognized as a hepatic manifestation of MS [4][34] which shares a common pathogenic pathway in insulin resistance. The pathophysiology involves an imbalance between lipid acquisition, mitochondrial fatty acid oxidation, and its export as part of the very-low-density lipoprotein (VLDL) molecule that generates hepatic steatosis [35]. In this way, it is clear that the criteria for MS (dyslipidemia, hyperglycemia, central adiposity, and hypertension) and weight gain will be considered risk factors strongly related to the disease [36].
In NASH, the two-hit proposal has been used for years to explain the pathophysiology of the disease. The first involves insulin resistance that will cause steatosis, and the second is associated with the inflammatory process generated by lipid oxidation. Nevertheless, both hits are insufficient to explain the disease. Thus, a multi-hit theory was proposed, including (besides the phenomena of the old theory) lipotoxicity caused by the accumulation of free fatty acids, cholesterol, and triglycerides, Kupffer cell activation, myeloid cell recruitment, gut microbiota dysfunction, genetic factors, and diet [35][37][38].
Recently, some authors have joined in favor of changing the name from NAFLD to metabolic (dysfunction) associated with fat liver disease (MAFLD). They believe that the term NAFLD has been described as an exclusionary condition. It exists only when other conditions such as viral hepatitis B and C, autoimmune diseases, or alcohol intake above a certain threshold are absent. However, MAFLD is present in about one fourth of the global population, and it coexists with other liver diseases. Another argument resides in the debate about the safe limit of alcohol intake and the challenge about the application of questionnaires that are faithful to the real consumption of this beverage. The third point is that the new term could simplify the stratification of the disease without the dichotomous classification between NASH and non-NASH. Finally, the authors argue that the new term would consider the heterogeneity of fat liver disease which facilitates the selection of phenotypes for clinical trials as well as therapies. The diagnosis of MAFLD is grounded on the identification of hepatic steatosis by histology, imaging, or blood biomarkers, in association with one of the following three conditions: excess adiposity, presence of pre-diabetes or DM2, or evidence of metabolic dysregulation [39][40][41][42]. Figure 1 shows the relationship between the gut microbiome, NAFLD, and NASH.
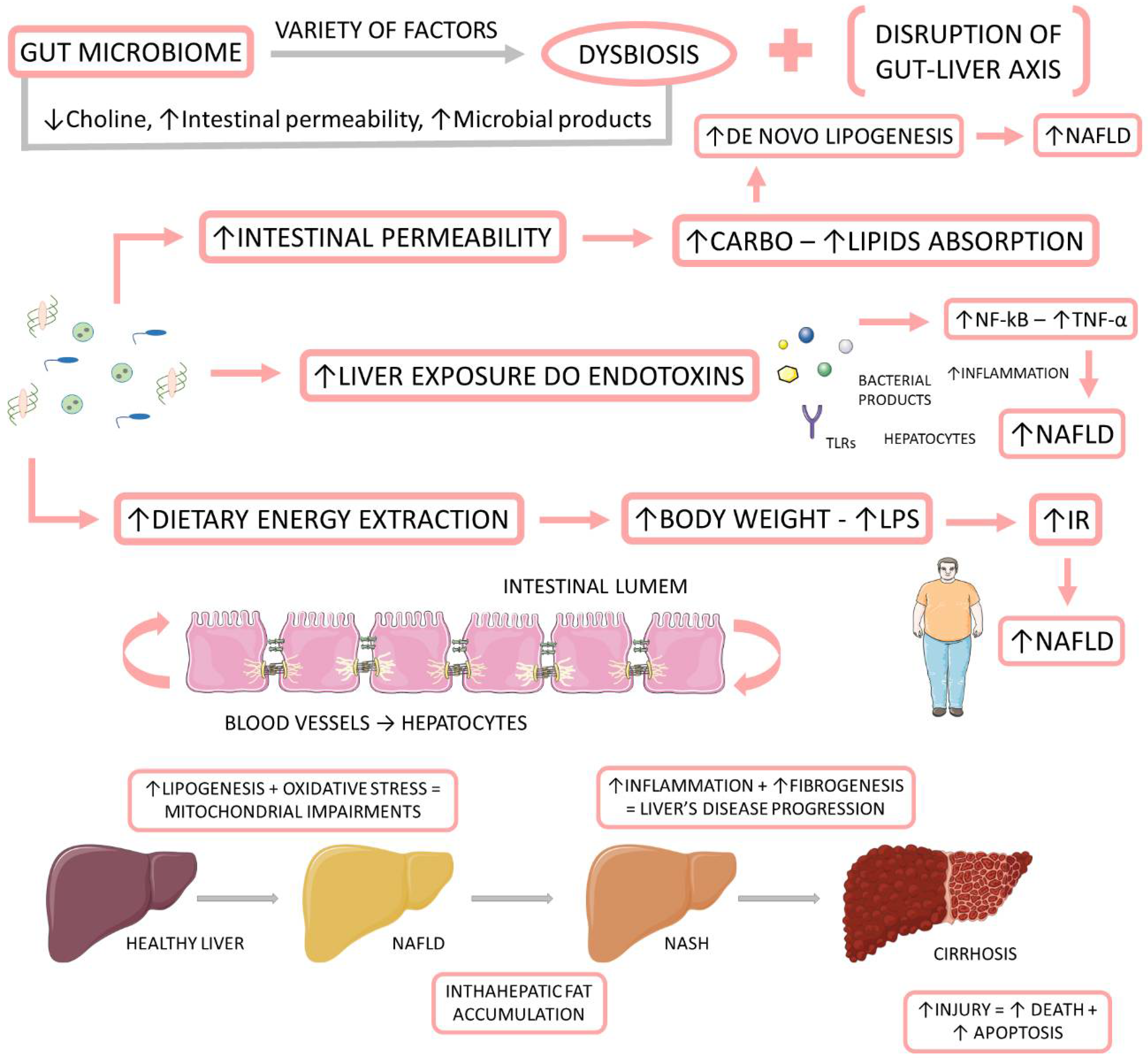
Figure 1. The relationship between the gut microbiome, NAFLD, and NASH. The gut microbiome is affected by a variety of factors to be in dysbiosis. When dysbiotic, the gut microbiome becomes disrupted and starts to cause alterations in the intestinal permeability, leading to augmented liver exposure to endotoxins and dietary energy extraction. These alterations induce an increase in the intrahepatic lipid accumulation, and they induce liver inflammation and fibrosis. IR: insulin resistance; LPS: Lipopolysaccharide; NF-kB: nuclear factor kappa B; NAFLD: non-alcoholic fatty liver disease; NASH: non-alcoholic steatohepatitis; TNF-α: tumor necrosis factor-alpha.
3. Microbiome
The human microbiome refers to the genomic component of organisms (microbiota) that inhabit a specific human body location [43]. More than 30 trillion microorganisms are part of this ecosystem [44]. It is composed mainly of bacteria, but it includes commensal populations of fungi, archaea, and protists as well, covering all three domains (Bacteria, Archaea, and Eukarya), in addition to viruses [45][46]. These microbes reside in the skin, oral cavity, gastrointestinal, respiratory, and genitourinary tracts, accounting for 1–3% of total body weight [47]. Considering the variability between individuals, the intestinal microbiome has a set of 3.3 million different genes, representing a genome 150 times larger than humans’ [48].
The intestinal microbiota plays a vital role in the metabolism of substrates including carbohydrates, proteins, polyphenols, vitamins, and bile [48][49]. It is closely related to the hosts which develop and tune the immune system [50], as well as protect against pathogenic colonization by competing for fixation sites or nutrient sources, producing bacteriocins (e.g., lactic acid), inhibitory metabolites (short-chain fatty acids and lithocholic acid), stimulating IgA epithelization and mucus production [51][52]. Moreover, an interesting relationship between microbiota and the nervous system has been observed, constituting the brain–gut axis (Figure 2). This extensive communication network connects the gastrointestinal tract with the central nervous system’s cognitive and emotional centers (CNS) [53][54].
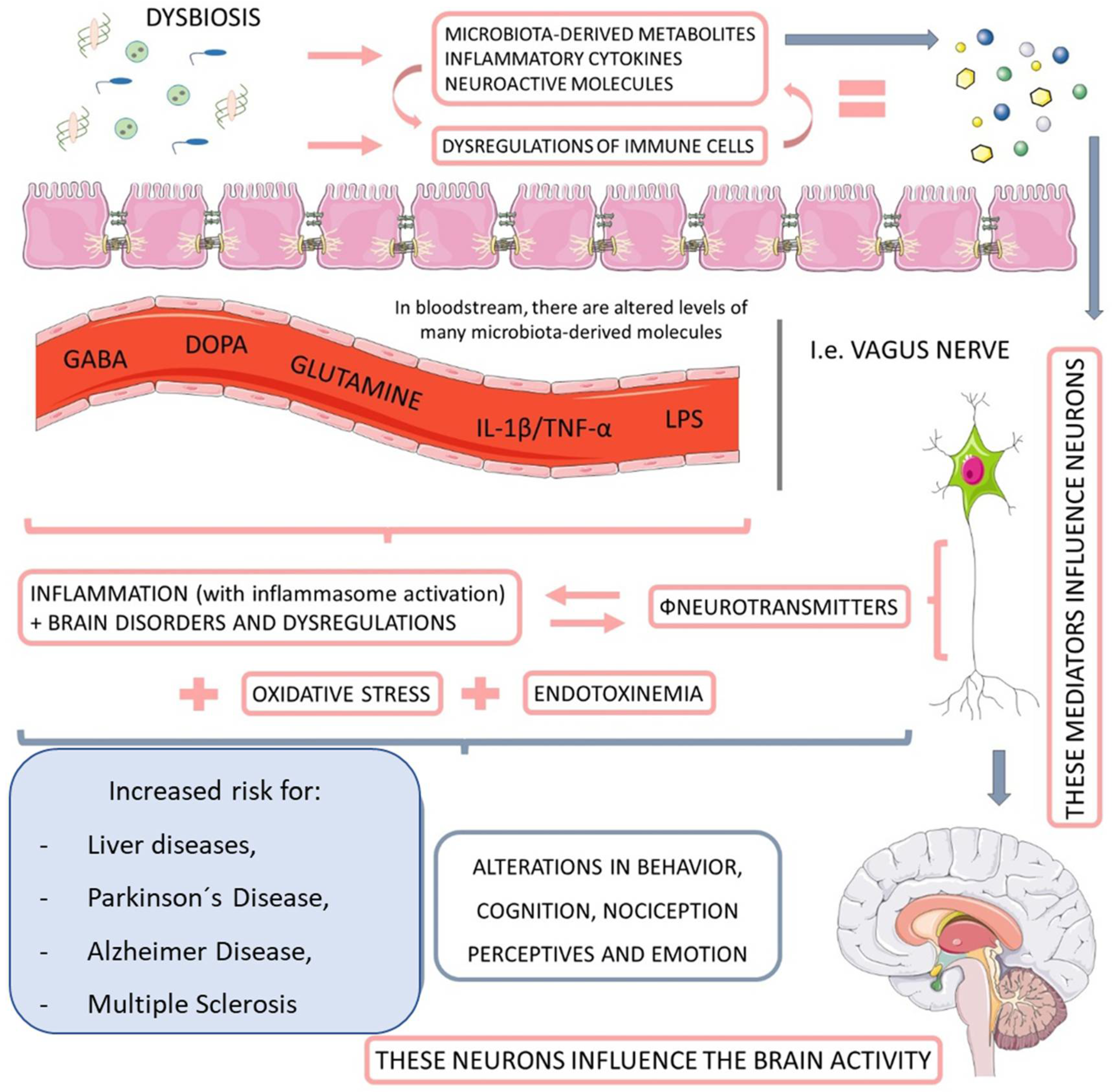
Figure 2. The relationship between microbiota, nervous system, and liver diseases. When dysbiotic, the gut-microbiome-derived metabolites start to cause neuroendocrine dysregulation, principally by impairments in the activity of neurotransmitters. This endotoxemia increases the personal risk for Parkinson’s disease and Alzheimer’s disease, as well as multiple sclerosis and other behavioral and cognitional alterations. GABA: gamma-Aminobutyric Acid; DOPA: dopamine; IL-1ß: interleukin-1β; TNF-α: tumor necrosis factor.
It was established that the fetus’s intrauterine environment was sterile for a long time, and the microbiome development started during and after birth [55]. However, in the last 15 years, with advances in DNA sequencing technology, this theory has been challenged by scientific evidences that demonstrated the presence of microbes in the placenta, amniotic fluid, the umbilical blood cord, and meconium (even in healthy pregnancies), [55][56][57][58][59][60][61] in addition to the possibility of participation of maternal microbiota and its metabolites in fetal development, inciting the theory of in utero colonization [62].
During and after birth, the newborn is surrounded by many microorganisms. Thus, the development and shaping of the initial microbiota will depend on and vary according to factors such as gestational time, mode of delivery, method of infant feeding, intrapartum, and neonatal antibiotic courses [63]. Neonates born from normal births are likely to have fecal microbiota resembling vaginal microbiota, dominated by Prevotella spp. and Lactobacillus, while babies born by cesarean section acquire bacteria derived from the hospital environment and maternal skin, such as Staphylococcus, Corynebacterium, Propionibacterium spp. [64]. This differentiation is relevant as neonates born by cesarean section are more susceptible to developing asthma, rhinitis, food allergy, celiac disease, and overweightness over the years [65]. The mode of infant feeding has a substantial influence because breast milk is rich in human milk oligosaccharides (HMOs) which are involved in pathogenic protection, maturation of the intestinal microbiome, and promotion of intestinal barrier function and maturation of immune cells [66]. Bifidobacterium bacteria are the most related to the positive effects of HMOs and are generally the most abundant among taxons found in infant intestinal microbiota (up to 90%) [67] that have their production two times higher in breastfed newborns compared to feed formulas [68]. Gestation time is also essential, as premature neonates’ immature intestines may have peristalsis, poor barrier functions, and immunity, which may precede the onset of infection and the inflammatory process by colonization pathogenic bacteria [69]. Although decreasing the colonization of pathogenic bacteria, intrapartum antibiotics administration was also related to the development of necrotizing enterocolitis (NEC). Besides, antibiotic administration in the first six months was correlated with an increased likelihood of developing asthma and obesity [70]. Other factors such as family environment, geographical, and cultural traditions are also documented as influencing infant microbiota [63].
In summary, the first colonizers of the newborn’s intestinal microbiota are usually facultative anaerobes, followed by the accumulation of obligatory anaerobes, including Bifidobacterium, Bacteroides, and Clostridium for the following six months [71]. With weaning and the introduction of solid foods from six months onwards, intestinal microbiota diversity increases, with Actinobacteria and Proteobacteria becoming the dominant components of infant microbiota [72]. At the age of 2.5, the infant microbiota’s composition, diversity, and functional capabilities resemble adult microbiota [73]. This intestinal community will undergo subtle changes until middle age (around 40 years of age), which is a time of relative stability [74]. It has a robust interpersonal character but is generally dominated by Firmicutes and Bacteroidetes, representing up to 90% of its composition and Actinobacteria, Proteobacteria, and Verrucomicrobia phyla [75].
However, even in stable chronological periods, the microbiota is subject to conditions capable of destabilizing homeostasis with the host. This process is called dysbiosis, a compositional and functional change in the microbiota that is driven by a set of factors that disturb the microbial ecosystem to a certain extent that exceeds its capacity for resistance and resilience. Several factors are associated with this phenomenon, including infections, diet, xenobiotics, genetics, familial transmission, circadian disruption, high-fat maternal diet, pregnancy, and physical injury [76].
An ecosystem in dysbiosis can damage the host immune system through various mechanisms that collectively stabilize the dysbiotic configuration. These mechanisms include modulation of inflammatory signaling by microbial metabolites, modulation of Toll-like receptor signaling (TLR), and degradation of IgA secreting agent (sIgA). The result is an intestinal epithelium more susceptible to pathobionts and disruption of the immune system, which decreases the protective capacity of the intestinal barrier, stimulating local or systemic inflammatory and immune-mediated processes [76][77][78]. Therefore, the genesis of several diseases had already been related to the microbiota’s dysbiotic configuration, including DM2, obesity, inflammatory bowel disease, cardiovascular diseases, autoimmune diseases, neurodegenerative diseases, NAFLD and its progressive form, non-alcoholic steatohepatitis (NASH) [79][80][81][82][83][84][85].
4. Microbiome, NAFLD, and NASH
4.1. Associations between the Gut Microbiome and NAFLD and NASH
The reciprocal interaction between the microbiome and the liver is established through the vascular route of the portal vein that takes to the liver gut-derived products and the liver feedback route of bile and antibody secretion to the intestine [15]. The liver is first exposed to gut-derived toxic factors, including bacteria, damaged metabolites, or bacterial products (LPS and bacteria DNA) [86].
Le roy et al. [87] demonstrated that when exposed to a high-fat diet, germ-free (GF) rats that received a transfer of gut microbiota from hyperglycaemic rats developed fasting hyperglycemia, hyperinsulinemia, and hepatic macrovesicular steatosis. In contrast, GF rats that received a transfer from normoglycemic rats remained normoglycaemic and without steatosis. Pyrosequencing of the 16S ribosomal RNA genes showed that hyperglycemic and normoglycemic rats had distinct gut microbiota regarding phylum, genus, and species. Henao-Mejia et al. [88] showed that sharing the microbiota through coprophagy from mice prone to developing NASH due to modifications in the inflammasome pathway in wild mice led to the development of steatosis and inflammation in the latter group.
Interestingly, some studies have observed that each stage of NAFLD corresponds to a pattern of gut microbiota [89]. A prospective study demonstrated that specific bacterial metagenomic signatures in the gut microbiome of NAFLD patients are a robust predictor of advanced fibrosis in humans. Some species were associated with NAFLD, with the abundance of bacterial species, such as Proteobacteria, Enterobacteria, Escherichia, and Bacteroides, being higher in NASH patients compared to matched healthy subjects [90].
There are several mechanisms by which the intestinal microbiota interfere in the progression of NAFLD and NASH. The increase in intestinal permeability, the translocation of dysbiotic bacteria, and the production of metabolites can be associated due to this dysbiotic state, and it is capable of generating disordered inflammatory responses which influence liver metabolism [91].
A new therapeutic attempt that has recently emerged is fecal microbiota transplantation. In this regard, Craven et al. [92] compared two groups: one received an autologous FMT, and the other received allogeneic FMT sourced from three lean, healthy individuals. At the end of the research, there was a significant decrease in the small intestinal permeability of the allogeneic group compared with the autologous group. Although the research did not show changes on biomarkers, it is known that increased intestinal permeability represents a central mechanism behind diseases such as inflammatory bowel disease, systemic inflammation, infection, MS, and NAFLD.
4.2. Microbiota-Derived Metabolites and Their Impact on NAFLD and NASH
Short-Chain Fatty Acids (SCFAs)
The short-chain fatty acids (SCFA) are the primary end products of fermentation of nondigestible carbohydrates (NDC) that become available to the gut microbiota. The main NDC are acetate, propionate, and butyrate [93][94]. Bacteroides are the main producers of propionate and acetate, while Firmicutes are the primary producers of butyrate [95]. Butyrate and propionate are well documented as gut inflammation relievers [96]. In rats, acetate and propionate supplementation decreased lipogenesis and fat accumulation, shielding them from high-fat (HF) diet-induced weight gain [97]. Svegliati-Baroni et al. [98] reported that the expression of the glucagon-like peptide-1 receptor (GLP-1r) is reduced in the hepatocytes of rats fed diet HF and patients with NASH, and that the activation of GLP-1r in the hepatocytes increased the oxidation of β-fatty acids and improved the insulin sensitivity. In addition, butyrate in particular is able to improve the function of tight junctions and stimulate mucin production, which helps maintain the integrity of the intestinal wall and prevent translocation of bacteria and its products, such as LPS, into the portal circulation [99][100].
Bile Acids
Bile acids are molecules produced in the liver from cholesterol and stored in the gallbladder. In addition to facilitating the absorption of lipids, they also play a role in glucose metabolism. The intestinal microbiota converts primary bile acids, including cholic acid (CA) and chenodeoxycholic acid (CDCA) in the distal small intestine and colon of humans into more than twenty different secondary bile acids, such as deoxycholic acid (DCA), lithocholic acid (LCA) and ursodeoxycholic acid (UDCA) [101][102]. Bile acids are indirectly involved in antimicrobial defense mediated by the farnesoid X receptor (FXR). Activation of this receptor reduces fatty acid and triglyceride synthesis in the liver by decreasing the expression of LXR and SREBP-1C [103]. FXR-deficient mice show reduced insulin sensitivity and decreased glucose tolerance [104]. In contrast, FXR activation by selective agonists suppresses bile acid and fatty acid production and increases glucose and insulin sensitivity in obese and diabetic mice. FXR activation also appears to attenuate primary biliary cirrhosis and NASH by reducing bile acid pool and liver fibrosis [105][106]. Bile acids are also closely related to another receptor, Takeda-G-protein-receptor-5 (TGR5). In the intestines, activation of TGR5 on L cells increases secretion of GLP-1, which binds to its receptor located on pancreatic beta cells, raising insulin secretion and reducing glucagon synthesis. The TGR5 is also able to modulate inflammatory processes. Its binding to the receptor reduces the release of pro-inflammatory cytokines by macrophages through the inhibition of NF-kB. Furthermore, in an experimental animal model, TGR5 knockout (TGR5-/-) mice have been shown to display accelerated LPS-induced inflammation in the liver and to suppress the inhibitory effect of TGR5 agonist on the expression of inflammatory mediators when compared with wild-type mice [107].
Choline and Trimethylamine
Choline is a constituent of the cells and mitochondrial membranes and the neurotransmitter acetylcholine. Choline-deficient diets have long been used to examine the mechanisms of fatty liver disease and its progression. They reproduce many of the phenotypes seen in humans with NAFLD, including an accumulation of triglycerides in the liver [108]. The phosphorylation of choline for the production of phospholipids and its oxidation as a methyl group donor are the main destinations of this nutrient [109]. Phosphatidylcholine is one of the most important choline metabolites. Its function is related to the packaging and export of triglycerides in very-low-density lipoprotein (VLDL) and to the solubilization of bile acids for excretion [110][111]. The lack of choline alters mitochondrial membranes, decreasing the concentration of phosphatidylethanolamine and phosphatidylcholine, which leads to a decrease in an action potential. This process consequently decreases ATP production and beta-oxidation, causing further hepatic steatosis [112][113]. For example, Arao et al. [114] used a methionine/choline-deficient diet to establish a NASH model and found that mitochondrial DNA content was decreased. The gut microbiome actively metabolizes choline, which may alter its bioavailability and potentially predispose one to choline deficiency [115]. The intestinal microbiota promotes the conversion of choline into trimethylamine, which, upon entering the circulation, will be converted into trimethylamine N-oxide in the liver [116]. Increased production of this substance results in a decrease in choline and consequently in the export of hepatic very-low-density lipoproteins and modulation of bile acid synthesis, which has detrimental effects on the liver, such as increased hepatic fat deposition and inflammatory and oxidative lesions and decreased glucose metabolism [89][117].
Ethanol
Ethanol is a microbial metabolite derived from saccharolytic fermentation. As late as 2000, Cope et al. [118] suggested that blood levels of ethanol were related to changes in the gut microbiota. Further, other studies have shown that dysbiosis in NASH patients involves ethanol-producing bacteria such as Escherchia coli, Bacteroides, Bifidobacterium, Clostridium [119][120]. One survey found a high increase in ethanol levels in NASH patients compared to healthy individuals or obese non-NASH patients [119]. Gut-bacteria-derived ethanol and its oxidized metabolite, acetaldehyde, are possibly involved in the progression of NAFLD through direct toxic effects on liver cells, through damage to the intestinal barrier generating increased portal endotoxemia, and through upregulation of nuclear factor-κB (NF-κB) signaling inflammatory pathways in peripheral cells [121][122]. Figure 3 shows the microbiota-derived metabolites and their impact on NAFLD and NASH.
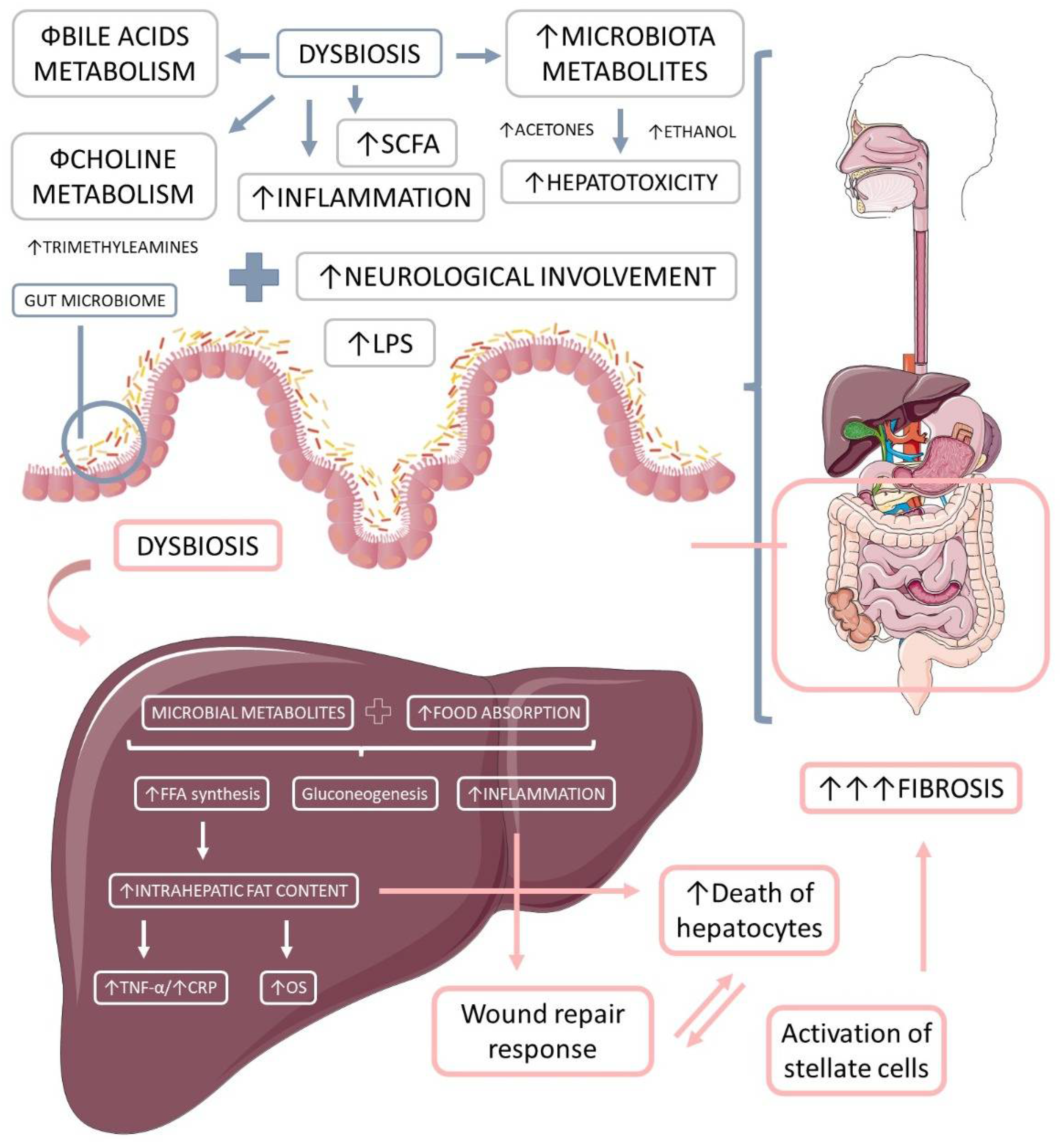
Figure 3. The microbiota-derived metabolites and their impact on liver inflammation, oxidative stress, and the development of liver diseases. Gut microbiome dysbiosis impairs the bile acids and choline metabolism, increases hepatotoxicity, and promotes inflammation. The microbiota-derived metabolites and the augmented food absorptions increase the intrahepatic production and accumulation of lipids, which causes increased inflammation and oxidative stress. Due to these events, the liver loses its capacity for wound repair response, and in addition to the augmented hepatocytes death and the augmented activation of stellate cells, liver massive fibrosis occurs. LPS: lipopolysaccharide; OS: oxidative stress, TNF-α: tumor necrosis factor-alpha; FFA: free fat acids; CRP: C reactive protein.
References
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84.
- Kumar, R.; Priyadarshi, R.N.; Anand, U. Non-alcoholic Fatty Liver Disease: Growing Burden, Adverse Outcomes and Associations. J. Clin. Transl. Hepatol. 2020, 8, 76–86.
- Hydes, T.J.; Summers, N.; Brown, E.; Alam, U.; Thomaides-Brears, H.; Wilding, J.P.H.; Cuthbertson, D.J. Mechanisms, screening modalities and treatment options for individuals with non-alcoholic fatty liver disease and type 2 diabetes. Diabet Med. 2020, 37, 1793–1806.
- Lin, H.; Yip, T.C.; Zhang, X.; Li, G.; Tse, Y.K.; Hui, V.W.; Liang, L.Y.; Lai, J.C.; Chan, S.L.; Chan, H.L.; et al. Age and the relative importance of liver-related deaths in nonalcoholic fatty liver disease. Hepatology 2022.
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Charlton, M.; Cusi, K.; Rinella, M.; Harrison, S.A.; Brunt, E.M.; Sanyal, A.J. The diagnosis and management of nonalcoholic fatty liver disease: Practice guidance from the American Association for the Study of Liver Diseases. Hepatology 2018, 67, 328–357.
- Ajmera, V.; Kim, B.K.; Yang, K.; Majzoub, A.M.; Nayfeh, T.; Tamaki, N.; Izumi, N.; Nakajima, A.; Idilman, R.; Gumussoy, M.; et al. Liver Stiffness on Magnetic Resonance Elastography and the MEFIB Index and Liver-Related Outcomes in Nonalcoholic Fatty Liver Disease: A Systematic Review and Meta-Analysis of Individual Participants. Gastroenterology 2022.
- Cen, J.; Han, Y.; Liu, Y.; Hu, H. Evaluated Glomerular Filtration Rate Is Associated with Non-alcoholic Fatty Liver Disease: A 5-Year Longitudinal Cohort Study in Chinese Non-obese People. Front. Nutr. 2022, 9, 916704.
- Stine, J.G.; Wentworth, B.J.; Zimmet, A.; Rinella, M.E.; Loomba, R.; Caldwell, S.H.; Argo, C.K. Systematic review with meta-analysis: Risk of hepatocellular carcinoma in non-alcoholic steatohepatitis without cirrhosis compared to other liver diseases. Aliment. Pharmacol. Ther. 2018, 48, 696–703.
- Witkowski, M.; Moreno, S.I.; Fernandes, J.; Johansen, P.; Augusto, M.; Nair, S. The Economic Burden of Non-Alcoholic Steatohepatitis: A Systematic Review. PharmacoEconomics 2022, 40, 751–776.
- Sheka, A.C.; Adeyi, O.; Thompson, J.; Hameed, B.; Crawford, P.A.; Ikramuddin, S. Nonalcoholic Steatohepatitis: A Review. JAMA 2020, 323, 1175–1183.
- Paternostro, R.; Trauner, M. Current treatment of non-alcoholic fatty liver disease. J. Intern. Med. 2022, 292, 190–204.
- Wong, R.J.; Aguilar, M.; Cheung, R.; Perumpail, R.B.; Harrison, S.A.; Younossi, Z.M.; Ahmed, A. Nonalcoholic steatohepatitis is the second leading etiology of liver disease among adults awaiting liver transplantation in the United States. Gastroenterology 2015, 148, 547–555.
- Mahfood Haddad, T.; Hamdeh, S.; Kanmanthareddy, A.; Alla, V.M. Nonalcoholic fatty liver disease and the risk of clinical cardiovascular events: A systematic review and meta-analysis. Diabetes Metab. Syndr. 2017, 11, S209–S216.
- Sharpton, S.R.; Maraj, B.; Harding-Theobald, E.; Vittinghoff, E.; Terrault, N.A. Gut microbiome-targeted therapies in nonalcoholic fatty liver disease: A systematic review, meta-analysis, and meta-regression. Am. J. Clin. Nutr. 2019, 110, 139–149.
- Albillos, A.; de Gottardi, A.; Rescigno, M. The gut-liver axis in liver disease: Pathophysiological basis for therapy. J. Hepatol. 2020, 72, 558–577.
- Li, P.; Hu, J.; Zhao, H.; Feng, J.; Chai, B. Multi-Omics Reveals Inhibitory Effect of Baicalein on Non-Alcoholic Fatty Liver Disease in Mice. Front. Pharmacol. 2022, 13, 925349.
- Yao, N.; Yang, Y.; Li, X.; Wang, Y.; Guo, R.; Wang, X.; Li, J.; Xie, Z.; Li, B.; Cui, W. Effects of Dietary Nutrients on Fatty Liver Disease Associated With Metabolic Dysfunction (MAFLD): Based on the Intestinal-Hepatic Axis. Front. Nutr. 2022, 9, 906511.
- Owaki, T.; Kamimura, K.; Ko, M.; Nagayama, I.; Nagoya, T.; Shibata, O.; Oda, C.; Morita, S.; Kimura, A.; Sato, T.; et al. The liver-gut peripheral neural axis and nonalcoholic fatty liver disease pathologies via hepatic serotonin receptor 2A. Dis. Models Mech. 2022, 15, dmm049612.
- Pandey, K.; Naik, S.R.; Vakil, B.V. Probiotics, prebiotics and synbiotics-a review. J. Food Sci. Technol. 2015, 52, 7577–7587.
- Yadav, M.K.; Kumari, I.; Singh, B.; Sharma, K.K.; Tiwari, S.K.J.A.M. Probiotics, prebiotics and synbiotics: Safe options for next-generation therapeutics. Biotechnology 2022, 106, 505–521.
- Li, D.; Li, Y.; Yang, S.; Lu, J.; Jin, X.; Wu, M. Diet-gut microbiota-epigenetics in metabolic diseases: From mechanisms to therapeutics. Biomed. Pharmacother. Biomed. Pharmacother. 2022, 153, 113290.
- Musazadeh, V.; Roshanravan, N.; Dehghan, P.; Ahrabi, S.S. Effect of Probiotics on Liver Enzymes in Patients with Non-alcoholic Fatty Liver Disease: An Umbrella of Systematic Review and Meta-Analysis. Front. Nutr. 2022, 9, 844242.
- Kanchanasurakit, S.; Kositamongkol, C.; Lanoi, K.; Nunta, M.; Saetuan, T.; Chaiyakunapruk, N.; Saokaew, S.; Phisalprapa, P. Effects of Synbiotics, Probiotics, and Prebiotics on Liver Enzymes of Patients with Non-alcoholic Fatty Liver Disease: A Systematic Review and Network Meta-Analysis. Front. Nutr. 2022, 9, 880014.
- Sabirin, F.; Lim, S.M.; Neoh, C.F.; Ramasamy, K. Hepatoprotection of Probiotics against Non-Alcoholic Fatty Liver Disease in vivo: A Systematic Review. Front. Nutr. 2022, 9, 844374.
- Arellano-García, L.; Portillo, M.P.; Martínez, J.A.; Milton-Laskibar, I. Usefulness of Probiotics in the Management of NAFLD: Evidence and Involved Mechanisms of Action from Preclinical and Human Models. Int. J. Mol. Sci. 2022, 23, 3167.
- Huang, Y.; Wang, X.; Zhang, L.; Zheng, K.; Xiong, J.; Li, J.; Cong, C.; Gong, Z.; Mao, J. Effect of Probiotics Therapy on Nonalcoholic Fatty Liver Disease. Comput. Math. Methods Med. 2022, 2022, 7888076.
- Yang, R.; Shang, J.; Zhou, Y.; Liu, W.; Tian, Y.; Shang, H. Effects of probiotics on nonalcoholic fatty liver disease: A systematic review and meta-analysis. Expert Rev. Gastroenterol. Hepatol. 2021, 15, 1401–1409.
- Souza, C.A.; Rocha, R.; Costa, P.R.F.; Almeida, N.S.; Cotrim, H.P. Probiotic, Prebiotic or Symbiotic Supplementation Impacts on Intestinal Microbiota in Patients with Nonalcoholic Fatty Liver Disease: A Systematic Review. Arq. De Gastroenterol. 2022, 59, 123–128.
- Puri, P.; Sanyal, A.J. Nonalcoholic fatty liver disease: Definitions, risk factors, and workup. Clin. Liver Dis. 2012, 1, 99–103.
- Cariou, B.; Byrne, C.D.; Loomba, R.; Sanyal, A.J. Nonalcoholic fatty liver disease as a metabolic disease in humans: A literature review. Diabetes Obes. Metab. 2021, 23, 1069–1083.
- Natarajan, Y.; Patel, P.; Chu, J.; Yu, X.; Hernaez, R.; El-Serag, H.; Kanwal, F. Risk of Hepatocellular Carcinoma in Patients with Various HFE Genotypes. Dig. Dis. Sci. 2022.
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD development and therapeutic strategies. Nat. Med. 2018, 24, 908–922.
- Mundi, M.S.; Velapati, S.; Patel, J.; Kellogg, T.A.; Abu Dayyeh, B.K.; Hurt, R.T. Evolution of NAFLD and Its Management. Nutr. Clin. Pract. Off. Publ. Am. Soc. Parenter. Enter. Nutr. 2020, 35, 72–84.
- Loomba, R.; Sanyal, A.J. The global NAFLD epidemic. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 686–690.
- Manne, V.; Handa, P.; Kowdley, K.V. Pathophysiology of Nonalcoholic Fatty Liver Disease/Nonalcoholic Steatohepatitis. Clin. Liver Dis. 2018, 22, 23–37.
- Zelber-Sagi, S.; Lotan, R.; Shlomai, A.; Webb, M.; Harrari, G.; Buch, A.; Nitzan Kaluski, D.; Halpern, Z.; Oren, R. Predictors for incidence and remission of NAFLD in the general population during a seven-year prospective follow-up. J. Hepatol. 2012, 56, 1145–1151.
- Kořínková, L.; Pražienková, V.; Černá, L.; Karnošová, A.; Železná, B.; Kuneš, J.; Maletínská, L. Pathophysiology of NAFLD and NASH in Experimental Models: The Role of Food Intake Regulating Peptides. Front. Endocrinol. 2020, 11, 597583.
- Santos, J.; Maio, M.C.; Lemes, M.A.; Laurindo, L.F.; Haber, J.; Bechara, M.D.; Prado, P.S.D., Jr.; Rauen, E.C.; Costa, F.; Pereira, B.C.A.; et al. Non-Alcoholic Steatohepatitis (NASH) and Organokines: What Is Now and What Will Be in the Future. Int. J. Mol. Sci. 2022, 23, 498.
- Eslam, M.; Newsome, P.N.; Sarin, S.K.; Anstee, Q.M.; Targher, G.; Romero-Gomez, M.; Zelber-Sagi, S.; Wai-Sun Wong, V.; Dufour, J.F.; Schattenberg, J.M.; et al. A new definition for metabolic dysfunction-associated fatty liver disease: An international expert consensus statement. J. Hepatol. 2020, 73, 202–209.
- Liu, J.; Ayada, I.; Zhang, X.; Wang, L.; Li, Y.; Wen, T.; Ma, Z.; Bruno, M.J.; de Knegt, R.J.; Cao, W.; et al. Estimating Global Prevalence of Metabolic Dysfunction-Associated Fatty Liver Disease in Overweight or Obese Adults. Clin. Gastroenterol. Hepatol. Off. Clin. Pract. J. Am. Gastroenterol. Assoc. 2022, 20, e573–e582.
- Liu, J.; Mu, C.; Li, K.; Luo, H.; Liu, Y.; Li, Z. Estimating Global Prevalence of Metabolic Dysfunction-Associated Fatty Liver Disease in Overweight or Obese Children and Adolescents: Systematic Review and Meta-Analysis. Int. J. Public Health 2021, 66, 1604371.
- Méndez-Sánchez, N.; Bugianesi, E.; Gish, R.G.; Lammert, F.; Tilg, H.; Nguyen, M.H.; Sarin, S.K.; Fabrellas, N.; Zelber-Sagi, S.; Fan, J.G.; et al. Global multi-stakeholder endorsement of the MAFLD definition. Lancet. Gastroenterol. Hepatol. 2022, 7, 388–390.
- Ogunrinola, G.A.; Oyewale, J.O.; Oshamika, O.O.; Olasehinde, G.I. The Human Microbiome and Its Impacts on Health. Int. J. Microbiol. 2020, 2020, 8045646.
- Sender, R.; Fuchs, S.; Milo, R. Revised Estimates for the Number of Human and Bacteria Cells in the Body. PLoS Biol. 2016, 14, e1002533.
- Mills, S.; Stanton, C.; Lane, J.A.; Smith, G.J.; Ross, R.P. Precision Nutrition and the Microbiome, Part I: Current State of the Science. Nutrients 2019, 11, 923.
- Matijašić, M.; Meštrović, T.; Paljetak, H.; Perić, M.; Barešić, A.; Verbanac, D. Gut Microbiota beyond Bacteria-Mycobiome, Virome, Archaeome, and Eukaryotic Parasites in IBD. Int. J. Mol. Sci. 2020, 21, 2668.
- Ruan, W.; Engevik, M.A.; Spinler, J.K.; Versalovic, J. Healthy Human Gastrointestinal Microbiome: Composition and Function after a Decade of Exploration. Dig. Dis. Sci. 2020, 65, 695–705.
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T.; et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010, 464, 59–65.
- Rowland, I.; Gibson, G.; Heinken, A.; Scott, K.; Swann, J.; Thiele, I.; Tuohy, K. Gut microbiota functions: Metabolism of nutrients and other food components. Eur. J. Nutr. 2018, 57, 1–24.
- Belkaid, Y.; Harrison, O.J. Homeostatic Immunity and the Microbiota. Immunity 2017, 46, 562–576.
- Jandhyala, S.M.; Talukdar, R.; Subramanyam, C.; Vuyyuru, H.; Sasikala, M.; Nageshwar Reddy, D. Role of the normal gut microbiota. World J. Gastroenterol. 2015, 21, 8787–8803.
- Pickard, J.M.; Zeng, M.Y.; Caruso, R.; Núñez, G. Gut microbiota: Role in pathogen colonization, immune responses, and inflammatory disease. Immunol. Rev. 2017, 279, 70–89.
- Louwies, T.; Johnson, A.C.; Orock, A.; Yuan, T.; Greenwood-Van Meerveld, B. The microbiota-gut-brain axis: An emerging role for the epigenome. Exp. Biol. Med. 2020, 245, 138–145.
- Schaub, A.C.; Schneider, E.; Vazquez-Castellanos, J.F.; Schweinfurth, N.; Kettelhack, C.; Doll, J.P.K.; Yamanbaeva, G.; Mählmann, L.; Brand, S.; Beglinger, C.; et al. Clinical, gut microbial and neural effects of a probiotic add-on therapy in depressed patients: A randomized controlled trial. Transl. Psychiatry 2022, 12, 227.
- Perez-Muñoz, M.E.; Arrieta, M.C.; Ramer-Tait, A.E.; Walter, J. A critical assessment of the “sterile womb” and “in utero colonization” hypotheses: Implications for research on the pioneer infant microbiome. Microbiome 2017, 5, 48.
- Ardissone, A.N.; de la Cruz, D.M.; Davis-Richardson, A.G.; Rechcigl, K.T.; Li, N.; Drew, J.C.; Murgas-Torrazza, R.; Sharma, R.; Hudak, M.L.; Triplett, E.W.; et al. Meconium microbiome analysis identifies bacteria correlated with premature birth. PLoS ONE 2014, 9, e90784.
- Pelzer, E.; Gomez-Arango, L.F.; Barrett, H.L.; Nitert, M.D. Review: Maternal health and the placental microbiome. Placenta 2017, 54, 30–37.
- Collado, M.C.; Rautava, S.; Aakko, J.; Isolauri, E.; Salminen, S. Human gut colonisation may be initiated in utero by distinct microbial communities in the placenta and amniotic fluid. Sci. Rep. 2016, 6, 23129.
- Tapiainen, T.; Paalanne, N.; Tejesvi, M.V.; Koivusaari, P.; Korpela, K.; Pokka, T.; Salo, J.; Kaukola, T.; Pirttilä, A.M.; Uhari, M.; et al. Maternal influence on the fetal microbiome in a population-based study of the first-pass meconium. Pediatric Res. 2018, 84, 371–379.
- Walker, R.W.; Clemente, J.C.; Peter, I.; Loos, R.J.F. The prenatal gut microbiome: Are we colonized with bacteria in utero? Pediatric Obes. 2017, 12, 3–17.
- Schoenmakers, S.; Steegers-Theunissen, R.; Faas, M. The matter of the reproductive microbiome. Obstet. Med. 2019, 12, 107–115.
- Nyangahu, D.D.; Jaspan, H.B. Influence of maternal microbiota during pregnancy on infant immunity. Clin. Exp. Immunol. 2019, 198, 47–56.
- Butel, M.J.; Waligora-Dupriet, A.J.; Wydau-Dematteis, S. The developing gut microbiota and its consequences for health. J. Dev. Orig. Health Dis. 2018, 9, 590–597.
- Zhuang, L.; Chen, H.; Zhang, S.; Zhuang, J.; Li, Q.; Feng, Z. Intestinal Microbiota in Early Life and Its Implications on Childhood Health. Genom. Proteom. Bioinform. 2019, 17, 13–25.
- Montoya-Williams, D.; Lemas, D.J.; Spiryda, L.; Patel, K.; Carney, O.O.; Neu, J.; Carson, T.L. The Neonatal Microbiome and Its Partial Role in Mediating the Association between Birth by Cesarean Section and Adverse Pediatric Outcomes. Neonatology 2018, 114, 103–111.
- Hundshammer, C.; Minge, O. In Love with Shaping You-Influential Factors on the Breast Milk Content of Human Milk Oligosaccharides and Their Decisive Roles for Neonatal Development. Nutrients 2020, 12, 3568.
- Sakanaka, M.; Gotoh, A.; Yoshida, K.; Odamaki, T.; Koguchi, H.; Xiao, J.Z.; Kitaoka, M.; Katayama, T. Varied Pathways of Infant Gut-Associated Bifidobacterium to Assimilate Human Milk Oligosaccharides: Prevalence of the Gene Set and Its Correlation with Bifidobacteria-Rich Microbiota Formation. Nutrients 2019, 12, 71.
- Lewis, Z.T.; Sidamonidze, K.; Tsaturyan, V.; Tsereteli, D.; Khachidze, N.; Pepoyan, A.; Zhgenti, E.; Tevzadze, L.; Manvelyan, A.; Balayan, M.; et al. The Fecal Microbial Community of Breast-fed Infants from Armenia and Georgia. Sci. Rep. 2017, 7, 40932.
- Korpela, K.; Blakstad, E.W.; Moltu, S.J.; Strømmen, K.; Nakstad, B.; Rønnestad, A.E.; Brække, K.; Iversen, P.O.; Drevon, C.A.; de Vos, W. Intestinal microbiota development and gestational age in preterm neonates. Sci. Rep. 2018, 8, 2453.
- Zimmermann, P.; Curtis, N. Effect of intrapartum antibiotics on the intestinal microbiota of infants: A systematic review. Arch. Dis. Childhood. Fetal Neonatal. Ed. 2020, 105, 201–208.
- Robertson, R.C.; Manges, A.R.; Finlay, B.B.; Prendergast, A.J. The Human Microbiome and Child Growth–First 1000 Days and Beyond. Trends Microbiol. 2019, 27, 131–147.
- Rodríguez, J.M.; Murphy, K.; Stanton, C.; Ross, R.P.; Kober, O.I.; Juge, N.; Avershina, E.; Rudi, K.; Narbad, A.; Jenmalm, M.C.; et al. The composition of the gut microbiota throughout life, with an emphasis on early life. Microb. Ecol. Health Dis. 2015, 26, 26050.
- Thursby, E.; Juge, N. Introduction to the human gut microbiota. Biochem. J. 2017, 474, 1823–1836.
- De la Cuesta-Zuluaga, J.; Kelley, S.T.; Chen, Y.; Escobar, J.S.; Mueller, N.T.; Ley, R.E.; McDonald, D.; Huang, S.; Swafford, A.D.; Knight, R.; et al. Age- and Sex-Dependent Patterns of Gut Microbial Diversity in Human Adults. mSystems 2019, 4, 219–232.
- Illiano, P.; Brambilla, R.; Parolini, C. The mutual interplay of gut microbiota, diet and human disease. FEBS J. 2020, 287, 833–855.
- Levy, M.; Kolodziejczyk, A.A.; Thaiss, C.A.; Elinav, E. Dysbiosis and the immune system. Nat. Rev. Immunol. 2017, 17, 219–232.
- Weiss, G.A.; Hennet, T. Mechanisms and consequences of intestinal dysbiosis. Cell. Mol. Life Sci. CMLS 2017, 74, 2959–2977.
- Yoo, J.Y.; Groer, M.; Dutra, S.V.O.; Sarkar, A.; McSkimming, D.I. Gut Microbiota and Immune System Interactions. Microorganisms 2020, 8, 1587.
- Sikalidis, A.K.; Maykish, A. The Gut Microbiome and Type 2 Diabetes Mellitus: Discussing a Complex Relationship. Biomedicines 2020, 8, 8.
- Lee, C.J.; Sears, C.L.; Maruthur, N. Gut microbiome and its role in obesity and insulin resistance. Ann. N. Y. Acad. Sci. 2020, 1461, 37–52.
- Zuo, T.; Ng, S.C. The Gut Microbiota in the Pathogenesis and Therapeutics of Inflammatory Bowel Disease. Front. Microbiol. 2018, 9, 2247.
- Sanchez-Rodriguez, E.; Egea-Zorrilla, A.; Plaza-Díaz, J.; Aragón-Vela, J.; Muñoz-Quezada, S.; Tercedor-Sánchez, L.; Abadia-Molina, F. The Gut Microbiota and Its Implication in the Development of Atherosclerosis and Related Cardiovascular Diseases. Nutrients 2020, 12, 605.
- Jiao, Y.; Wu, L.; Huntington, N.D.; Zhang, X. Crosstalk Between Gut Microbiota and Innate Immunity and Its Implication in Autoimmune Diseases. Front. Immunol. 2020, 11, 282.
- Hirschberg, S.; Gisevius, B.; Duscha, A.; Haghikia, A. Implications of Diet and The Gut Microbiome in Neuroinflammatory and Neurodegenerative Diseases. Int. J. Mol. Sci. 2019, 20, 3109.
- Duarte, S.M.B.; Stefano, J.T.; Oliveira, C.P. Microbiota and nonalcoholic fatty liver disease/nonalcoholic steatohepatitis (NAFLD/NASH). Ann. Hepatol. 2019, 18, 416–421.
- Miele, L.; Marrone, G.; Lauritano, C.; Cefalo, C.; Gasbarrini, A.; Day, C.; Grieco, A. Gut-liver axis and microbiota in NAFLD: Insight pathophysiology for novel therapeutic target. Curr. Pharm. Des. 2013, 19, 5314–5324.
- Le Roy, T.; Llopis, M.; Lepage, P.; Bruneau, A.; Rabot, S.; Bevilacqua, C.; Martin, P.; Philippe, C.; Walker, F.; Bado, A.; et al. Intestinal microbiota determines development of non-alcoholic fatty liver disease in mice. Gut 2013, 62, 1787–1794.
- Henao-Mejia, J.; Elinav, E.; Jin, C.; Hao, L.; Mehal, W.Z.; Strowig, T.; Thaiss, C.A.; Kau, A.L.; Eisenbarth, S.C.; Jurczak, M.J.; et al. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature 2012, 482, 179–185.
- Chen, J.; Vitetta, L. Gut Microbiota Metabolites in NAFLD Pathogenesis and Therapeutic Implications. Int. J. Mol. Sci. 2020, 21, 5214.
- Loomba, R.; Seguritan, V.; Li, W.; Long, T.; Klitgord, N.; Bhatt, A.; Dulai, P.S.; Caussy, C.; Bettencourt, R.; Highlander, S.K.; et al. Gut Microbiome-Based Metagenomic Signature for Non-invasive Detection of Advanced Fibrosis in Human Nonalcoholic Fatty Liver Disease. Cell Metab. 2017, 25, 1054–1062.e5.
- Kolodziejczyk, A.A.; Zheng, D.; Shibolet, O.; Elinav, E. The role of the microbiome in NAFLD and NASH. EMBO Mol. Med. 2019, 11, e9302.
- Craven, L.; Rahman, A.; Nair Parvathy, S.; Beaton, M.; Silverman, J.; Qumosani, K.; Hramiak, I.; Hegele, R.; Joy, T.; Meddings, J.; et al. Allogenic Fecal Microbiota Transplantation in Patients with Nonalcoholic Fatty Liver Disease Improves Abnormal Small Intestinal Permeability: A Randomized Control Trial. Am. J. Gastroenterol. 2020, 115, 1055–1065.
- Den Besten, G.; van Eunen, K.; Groen, A.K.; Venema, K.; Reijngoud, D.J.; Bakker, B.M. The role of short-chain fatty acids in the interplay between diet, gut microbiota, and host energy metabolism. J. Lipid Res. 2013, 54, 2325–2340.
- Morrison, D.J.; Preston, T. Formation of short chain fatty acids by the gut microbiota and their impact on human metabolism. Gut Microbes 2016, 7, 189–200.
- Chakraborti, C.K. New-found link between microbiota and obesity. World J. Gastrointest. Pathophysiol. 2015, 6, 110–119.
- Ferreira, C.M.; Vieira, A.T.; Vinolo, M.A.; Oliveira, F.A.; Curi, R.; Martins Fdos, S. The central role of the gut microbiota in chronic inflammatory diseases. J. Immunol. Res. 2014, 2014, 689492.
- Weitkunat, K.; Stuhlmann, C.; Postel, A.; Rumberger, S.; Fankhänel, M.; Woting, A.; Petzke, K.J.; Gohlke, S.; Schulz, T.J.; Blaut, M.; et al. Short-chain fatty acids and inulin, but not guar gum, prevent diet-induced obesity and insulin resistance through differential mechanisms in mice. Sci. Rep. 2017, 7, 6109.
- Svegliati-Baroni, G.; Saccomanno, S.; Rychlicki, C.; Agostinelli, L.; De Minicis, S.; Candelaresi, C.; Faraci, G.; Pacetti, D.; Vivarelli, M.; Nicolini, D.; et al. Glucagon-like peptide-1 receptor activation stimulates hepatic lipid oxidation and restores hepatic signalling alteration induced by a high-fat diet in nonalcoholic steatohepatitis. Liver Int. Off. J. Int. Assoc. Study Liver 2011, 31, 1285–1297.
- Kelly, C.J.; Zheng, L.; Campbell, E.L.; Saeedi, B.; Scholz, C.C.; Bayless, A.J.; Wilson, K.E.; Glover, L.E.; Kominsky, D.J.; Magnuson, A.; et al. Crosstalk between Microbiota-Derived Short-Chain Fatty Acids and Intestinal Epithelial HIF Augments Tissue Barrier Function. Cell Host Microbe 2015, 17, 662–671.
- Wang, H.B.; Wang, P.Y.; Wang, X.; Wan, Y.L.; Liu, Y.C. Butyrate enhances intestinal epithelial barrier function via up-regulation of tight junction protein Claudin-1 transcription. Dig. Dis. Sci. 2012, 57, 3126–3135.
- Gérard, P. Metabolism of cholesterol and bile acids by the gut microbiota. Pathogens 2013, 3, 14–24.
- Wahlström, A.; Sayin, S.I.; Marschall, H.U.; Bäckhed, F. Intestinal Crosstalk between Bile Acids and Microbiota and Its Impact on Host Metabolism. Cell Metab. 2016, 24, 41–50.
- Yang, Z.X.; Shen, W.; Sun, H. Effects of nuclear receptor FXR on the regulation of liver lipid metabolism in patients with non-alcoholic fatty liver disease. Hepatol. Int. 2010, 4, 741–748.
- Ma, K.; Saha, P.K.; Chan, L.; Moore, D.D. Farnesoid X receptor is essential for normal glucose homeostasis. J. Clin. Investig. 2006, 116, 1102–1109.
- De Wit, N.; Derrien, M.; Bosch-Vermeulen, H.; Oosterink, E.; Keshtkar, S.; Duval, C.; de Vogel-van den Bosch, J.; Kleerebezem, M.; Müller, M.; van der Meer, R. Saturated fat stimulates obesity and hepatic steatosis and affects gut microbiota composition by an enhanced overflow of dietary fat to the distal intestine. Am. J. Physiology Gastrointest. Liver Physiol. 2012, 303, G589–G599.
- Hirschfield, G.M.; Mason, A.; Luketic, V.; Lindor, K.; Gordon, S.C.; Mayo, M.; Kowdley, K.V.; Vincent, C.; Bodhenheimer, H.C., Jr.; Parés, A.; et al. Efficacy of obeticholic acid in patients with primary biliary cirrhosis and inadequate response to ursodeoxycholic acid. Gastroenterology 2015, 148, 751–761.e8.
- Wang, Y.D.; Chen, W.D.; Yu, D.; Forman, B.M.; Huang, W. The G-protein-coupled bile acid receptor, Gpbar1 (TGR5), negatively regulates hepatic inflammatory response through antagonizing nuclear factor κ light-chain enhancer of activated B cells (NF-κB) in mice. Hepatology 2011, 54, 1421–1432.
- Hebbard, L.; George, J. Animal models of nonalcoholic fatty liver disease. Nat. Reviews. Gastroenterol. Hepatol. 2011, 8, 35–44.
- Corbin, K.D.; Zeisel, S.H. Choline metabolism provides novel insights into nonalcoholic fatty liver disease and its progression. Curr. Opin. Gastroenterol. 2012, 28, 159–165.
- Noga, A.A.; Vance, D.E. A gender-specific role for phosphatidylethanolamine N-methyltransferase-derived phosphatidylcholine in the regulation of plasma high density and very low density lipoproteins in mice. J. Biol. Chem. 2003, 278, 21851–21859.
- Li, Z.; Agellon, L.B.; Vance, D.E. Phosphatidylcholine homeostasis and liver failure. J. Biol. Chem. 2005, 280, 37798–37802.
- Teodoro, J.S.; Rolo, A.P.; Duarte, F.V.; Simões, A.M.; Palmeira, C.M. Differential alterations in mitochondrial function induced by a choline-deficient diet: Understanding fatty liver disease progression. Mitochondrion 2008, 8, 367–376.
- Serviddio, G.; Giudetti, A.M.; Bellanti, F.; Priore, P.; Rollo, T.; Tamborra, R.; Siculella, L.; Vendemiale, G.; Altomare, E.; Gnoni, G.V. Oxidation of hepatic carnitine palmitoyl transferase-I (CPT-I) impairs fatty acid beta-oxidation in rats fed a methionine-choline deficient diet. PLoS ONE 2011, 6, e24084.
- Arao, Y.; Kawai, H.; Kamimura, K.; Kobayashi, T.; Nakano, O.; Hayatsu, M.; Ushiki, T.; Terai, S. Effect of methionine/choline-deficient diet and high-fat diet-induced steatohepatitis on mitochondrial homeostasis in mice. Biochem. Biophys. Res. Commun. 2020, 527, 365–371.
- Romano, K.A.; Vivas, E.I.; Amador-Noguez, D.; Rey, F.E. Intestinal microbiota composition modulates choline bioavailability from diet and accumulation of the proatherogenic metabolite trimethylamine-N-oxide. mBio 2015, 6, e02481.
- Machado, M.V.; Cortez-Pinto, H. Diet, Microbiota, Obesity, and NAFLD: A Dangerous Quartet. Int. J. Mol. Sci. 2016, 17, 481.
- Dumas, M.E.; Barton, R.H.; Toye, A.; Cloarec, O.; Blancher, C.; Rothwell, A.; Fearnside, J.; Tatoud, R.; Blanc, V.; Lindon, J.C.; et al. Metabolic profiling reveals a contribution of gut microbiota to fatty liver phenotype in insulin-resistant mice. Proc. Natl. Acad. Sci. USA 2006, 103, 12511–12516.
- Cope, K.; Risby, T.; Diehl, A.M. Increased gastrointestinal ethanol production in obese mice: Implications for fatty liver disease pathogenesis. Gastroenterology 2000, 119, 1340–1347.
- Zhu, L.; Baker, S.S.; Gill, C.; Liu, W.; Alkhouri, R.; Baker, R.D.; Gill, S.R. Characterization of gut microbiomes in nonalcoholic steatohepatitis (NASH) patients: A connection between endogenous alcohol and NASH. Hepatology 2013, 57, 601–609.
- Chen, X.; Zhang, Z.; Li, H.; Zhao, J.; Wei, X.; Lin, W.; Zhao, X.; Jiang, A.; Yuan, J. Endogenous ethanol produced by intestinal bacteria induces mitochondrial dysfunction in non-alcoholic fatty liver disease. J. Gastroenterol. Hepatol. 2020, 35, 2009–2019.
- Baker, S.S.; Baker, R.D.; Liu, W.; Nowak, N.J.; Zhu, L. Role of alcohol metabolism in non-alcoholic steatohepatitis. PLoS ONE 2010, 5, e9570.
- Shen, Z.; Ajmo, J.M.; Rogers, C.Q.; Liang, X.; Le, L.; Murr, M.M.; Peng, Y.; You, M. Role of SIRT1 in regulation of LPS- or two ethanol metabolites-induced TNF-alpha production in cultured macrophage cell lines. Am. J. Physiology. Gastrointest. Liver Physiol. 2009, 296, G1047–G1053.
More
Information
Subjects:
Integrative & Complementary Medicine
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.4K
Entry Collection:
Gastrointestinal Disease
Revisions:
2 times
(View History)
Update Date:
16 Aug 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No