 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Carlos Del Pozo Losada | -- | 5752 | 2022-08-12 17:56:27 | | | |
| 2 | Vivi Li | Meta information modification | 5752 | 2022-08-15 04:09:25 | | |
Video Upload Options
The interest in 3,4-dihydropyrimidine-2(1H)-(thio)ones is increasing every day, mainly due to their paramount biological relevance. The Biginelli reaction is the classical approach to reaching these scaffolds, although the product diversity suffers from some limitations. In order to overcome these restrictions, two main approaches have been devised. The first one involves the modification of the conventional components of the Biginelli reaction and the second one refers to the postmodification of the Biginelli products. Both strategies have been extensively revised in this manuscript. Regarding the first one, initially, the modification of one of the components was covered. Although examples of modifications of the three of them were described, by far the modification of the keto ester counterpart was the most popular approach, and a wide variety of different enolizable carbonylic compounds were used; moreover, changes in two or the three components were also described, broadening the substitution of the final dihydropyrimidines. Together with these modifications, the use of Biginelli adducts as a starting point for further modification was also a very useful strategy to decorate the final heterocyclic structure.
1. Introduction
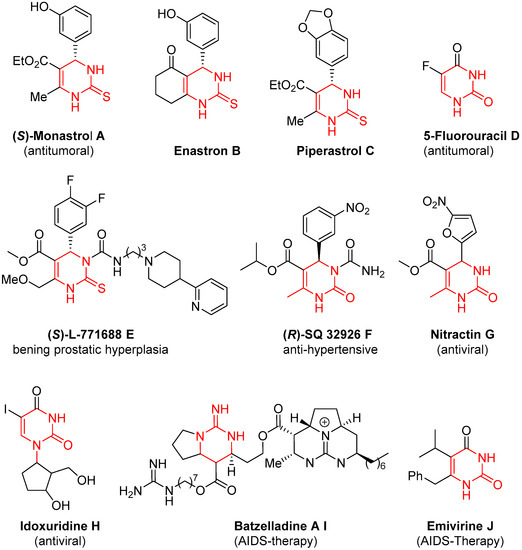
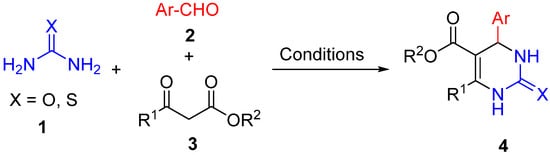
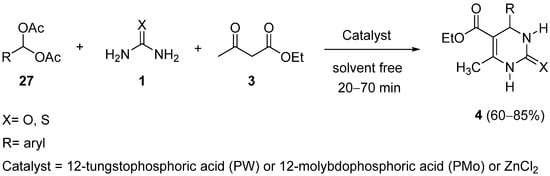
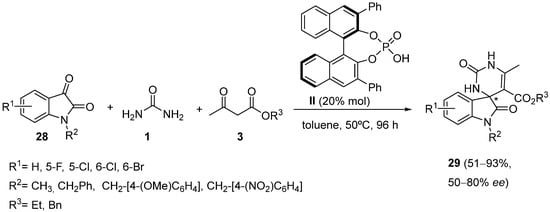
2. Modification of the Urea Counterpart
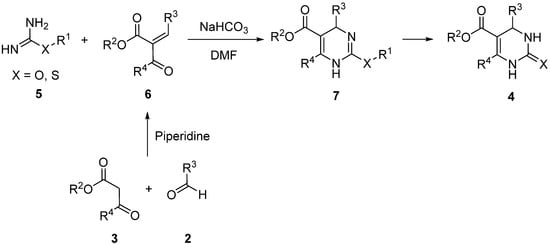
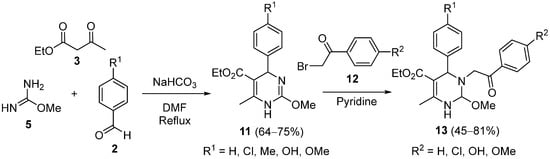
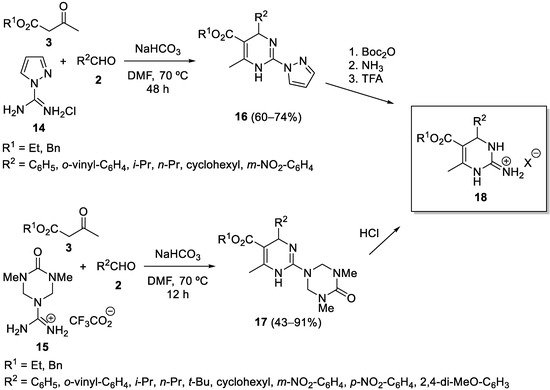
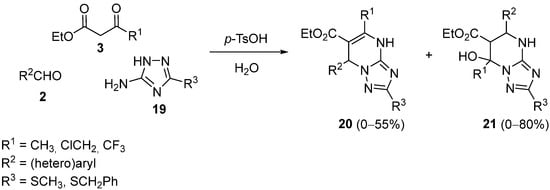
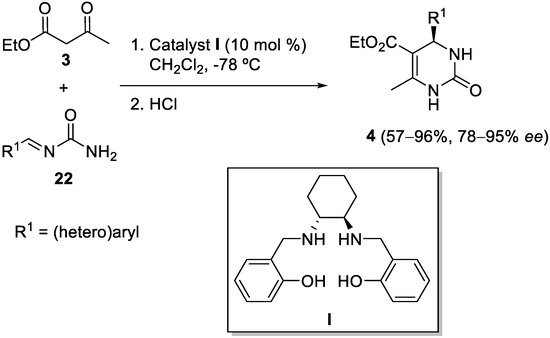
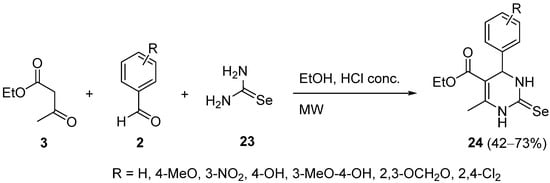
3. Modification of the Aldehyde Counterpart
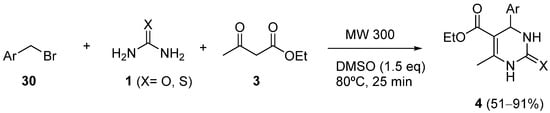
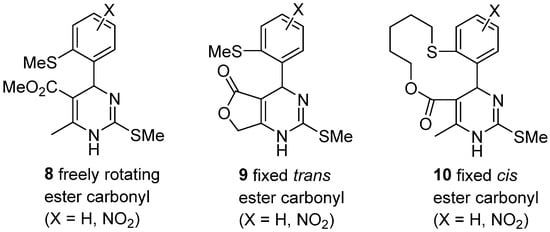
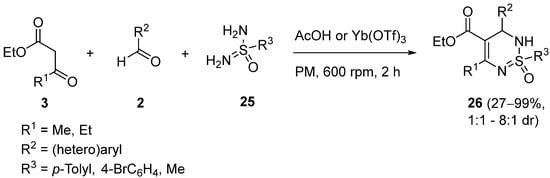
4. Modification of the Keto Ester Counterpart
4.1. Diketones as the Keto Ester Component
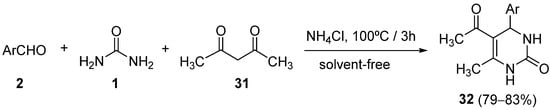
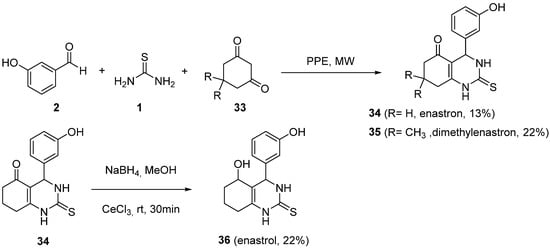
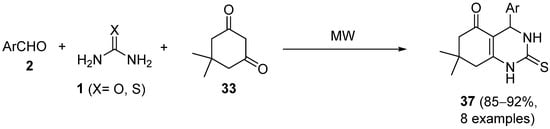
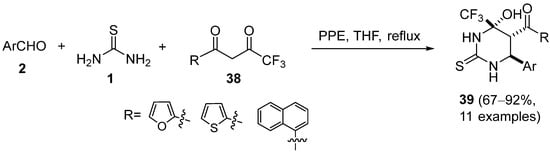
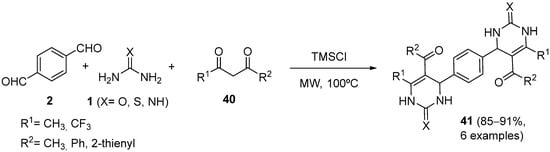
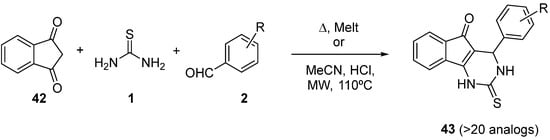
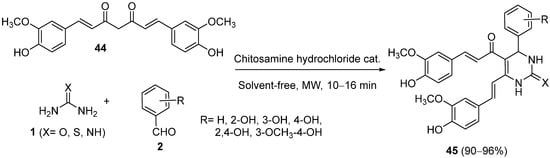
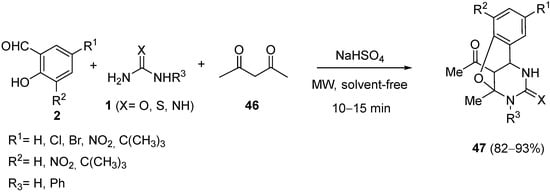
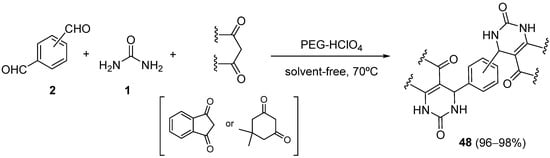
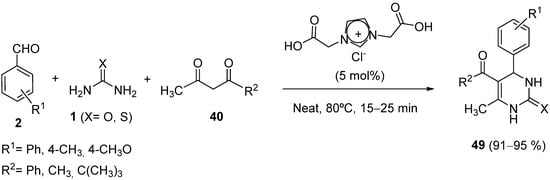
4.2. Keto Amides as the Keto Ester Component
4.2.1. Barbituric Acid Derivatives
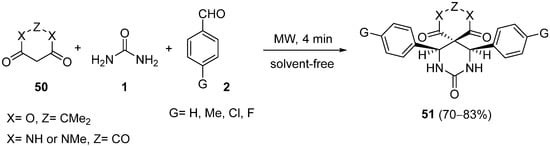
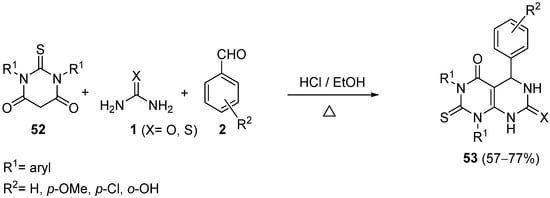
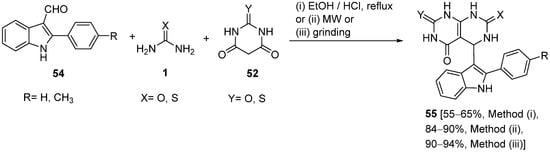
4.2.2. Beta-Ketoamides and Beta-Ketosulfonamides
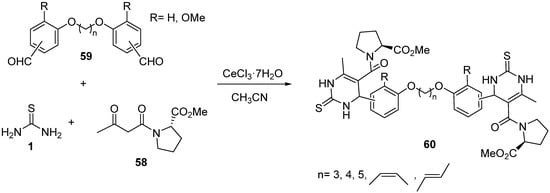
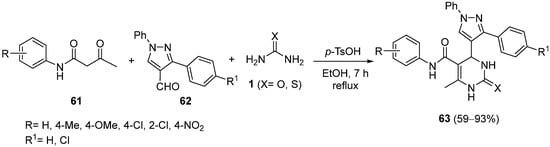
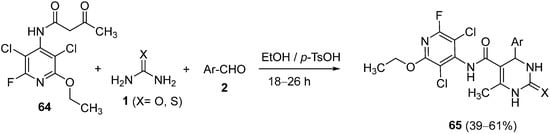
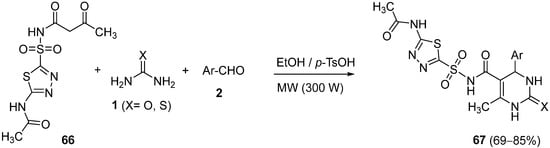
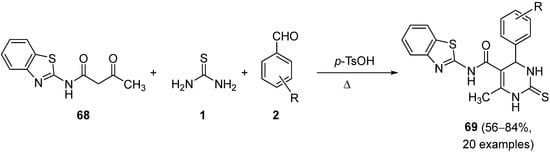
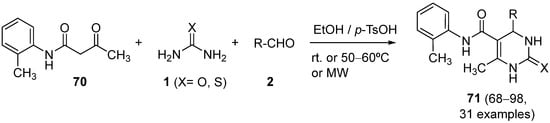
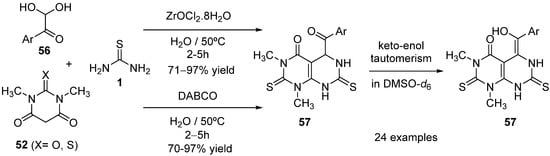
4.3. Keto Acids as the Keto Ester Component
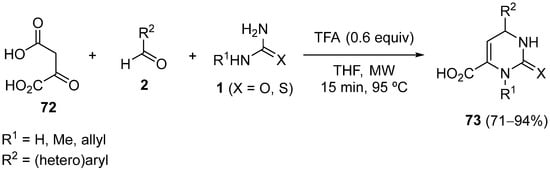
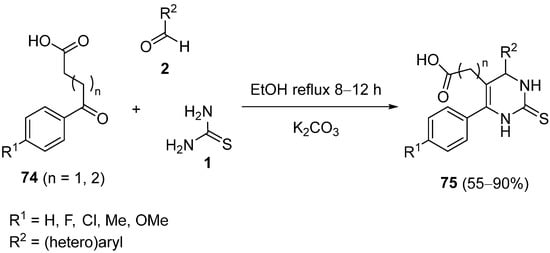
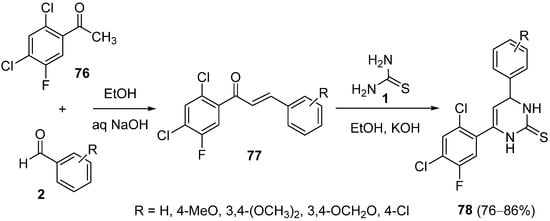
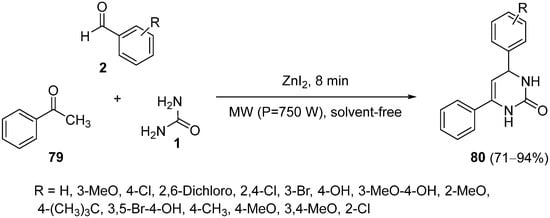
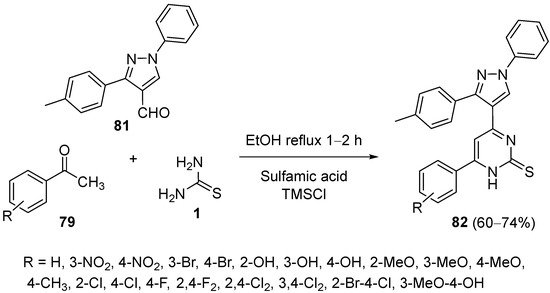
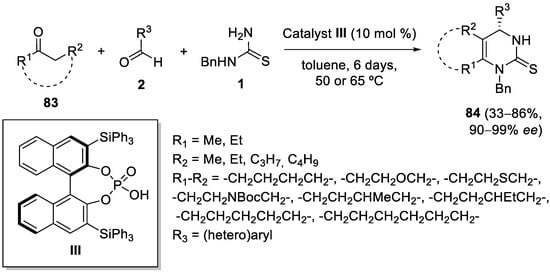
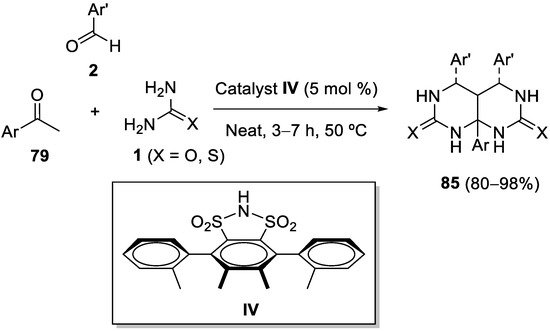
4.4. Ketones as the Keto Ester Component
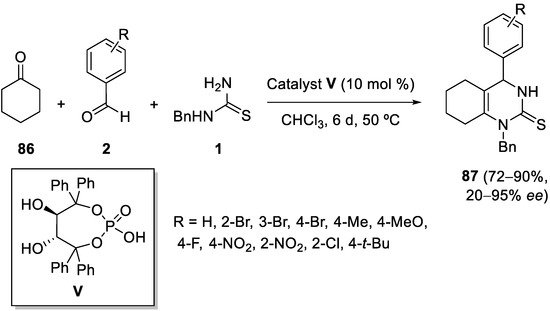
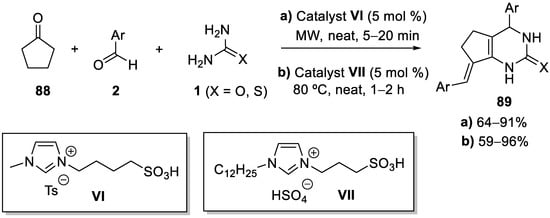
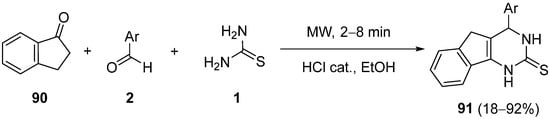
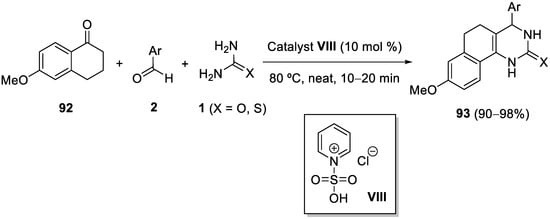
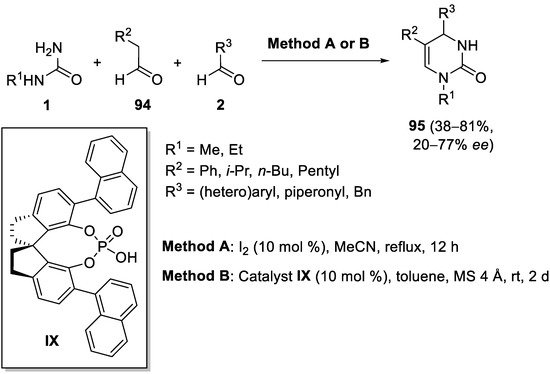
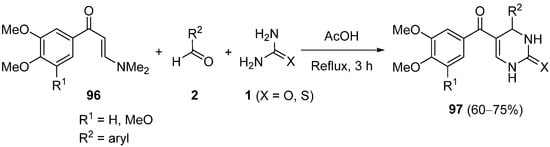
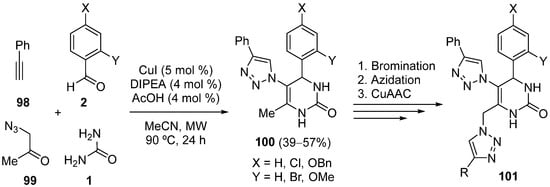
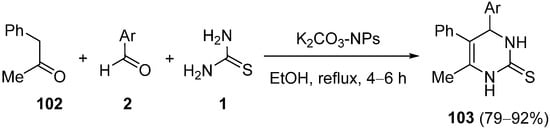
References
- Evans, B.E.; Rittle, K.E.; Bock, M.G.; DiPardo, R.M.; Freidinger, R.M.; Whitter, W.L.; Lundell, G.F.; Veber, D.F.; Anderson, P.S.; Chan, R.S.L.; et al. Methods for drug discovery development of potent, selective, orally effective cholecystokinin antagonists. J. Med. Chem. 1988, 31, 2235–2246.
- Patchett, A.A.; Nargund, R.P. Privileged structures—An update. Annual. Rep. Med. Chem. 2000, 35, 289–298.
- Khasimbi, S.; Ali, F.; Manda, K.; Sharma, A.; Chauhan, G.; Wakode, S. Dihydropyrimidinones scaffold as a promising nucleus for synthetic profile and various therapeutic targets: A Review. Curr. Org. Synth. 2021, 18, 270–293.
- Jeena, T.K.; George, M.; Joseph, L. A systematic review on the recent advances in the pharmacological activities of dihydropyrimidinones. Chem. Res. J. 2019, 4, 19–23.
- Santana-Matos, L.H.; Masson, F.T.; Simeoni, L.A.; Homem-de-Mello, M. Biological activity of dihydropyrimidinone (DHPM) derivatives: A systematic review. Eur. J. Med. Chem. 2018, 143, 1779–1789.
- Kaur, R.; Chaudhary, S.; Kumar, K.; Gupta, M.K.; Rawal, R.K. Recent synthetic and medicinal perspectives of dihydropyrimidinones: A review. Eur. J. Med. Chem. 2017, 132, 108–134.
- De Fátima, A.; Braga, T.C.; Neto, L.D.S.; Terra, B.S.; Oliveria, B.G.F.; da Silva, D.L.; Modolo, L.V. A mini-review on Biginelli adducts with notable pharmacological properties. J. Adv. Res. 2015, 6, 363–373.
- Wan, J.-P.; Pan, Y. Recent advance in the pharmacology of dihydropyrimidinone. Mini-Rev. Med. Chem. 2012, 12, 337–349.
- Suresh; Sandhu, J.S. Past, present and future of the Biginelli reaction: A critical perspective. ARKIVOC Online J. Org. Chem. 2011, 2012, 66–133.
- Kappe, C.O. Biologically active dihydropyrimidones of the Biginelli-type—A literature survey. Eur. J. Med. Chem. 2000, 35, 1043–1052.
- Adole, V.A. Synthetic approaches for the synthesis of dihydropyrimidinones/thiones (Biginelli adducts): A concise review. World. J. Pharm. Res. 2020, 9, 1067–1091.
- Suma, C.; Amritha, C.K.; Thushara, P.V.; Ananya, V.M. Dihydropyrimidinone derivatives—A review on synthesis and its therapeutic importance. Int. J. Pharm. Pharm. Res. 2020, 19, 271–286.
- Sharma, M.; Kaur, A. An update on the synthesis of dihydropyrimidinones using green chemistry approach. World J. Pharm. Pharm. Sci. 2016, 5, 949–967.
- Wan, J.-P.; Yunyun, L. Synthesis of dihydropyrimidinones and thiones by multicomponent reactions. Strategies beyond the classical Biginelli reaction. Synthesis 2010, 23, 3943–3953.
- Mayer, T.U.; Kapoor, T.M.; Haggarty, S.J.; King, R.W.; Schreiber, S.L.; Mitchison, T.J. Small molecule inhibitor of mitotic spindle bipolarity identified in a phenotype-based screen. Science 1999, 286, 971–974.
- Duschinsky, R.; Pleven, E.; Heidelberger, C. Synthesis of potential anticancer agents: 2-fluoroadenosine. J. Am. Chem. Soc. 1957, 79, 4559–4561.
- Barrow, J.C.; Nantermet, P.G.; Selnick, H.G.; Glass, K.L.; Rittle, K.E.; Gilbert, K.F.; Steele, T.G.; Homnick, C.F.; Freidinger, R.M.; Ransom, R.W.; et al. In vitro and in vivo evaluation of dihydropyrimidinone C-5 amides as potent and selective α1A receptor antagonists for the treatment of benign prostatic hyperplasia. J. Med. Chem. 2000, 43, 2703–2718.
- Atwal, K.S.; Swanson, B.N.; Unger, S.E.; Floyd, D.M.; Moreland, S.; Hedberg, A.; O´Reilly, B.C. Dihydropyrimidine Calcium Channel Blockers. 3-Carbamoyl-4-aryl-1,2,3,4-tetrahydro-6-methyl-5-pyrimidinecarboxylic Acid Esters as Orally Effective Antihypertensive Agents. J. Med. Chem. 1991, 34, 806–811.
- Hurst, E.W. Experimental chemotherapy of infection with agents of the psittacosis-lymphogranuloma-trachoma group. Ann. N. Y. Acad. Sci. 1962, 98, 275.
- Prusoff, W.H. Synthesis and biological activities of iododeoxyuridine, and analogue of thymidine. Biochim. Biophis. Acta 1959, 32, 295–296.
- Patil, A.D.; Kumar, N.V.; Kokke, W.C.; Bean, M.F.; Freyer, A.J.; De Brosse, C.; Mai, S.; Truneh, A.; Faulkner, D.J.; Carte, B.; et al. Novel alkaloids from the sponge Batzella sp.: Inhibitors of HIV gp120-human CD4 binding. J. Org. Chem. 1995, 60, 1182–1188.
- Hopkins, A.L.; Ren, J.; Esnouf, R.M.; Willcox, B.E.; Jones, E.Y.; Ross, C.; Miyasaka, T.; Walker, R.T.; Tanaka, H.; Stammers, D.K.; et al. Complexes of HIV-1 reverse transcriptase with inhibitors of the HEPT series reveal conformational changes relevant to the design of potent non-nucleoside inhibitors. J. Med. Chem. 1996, 39, 1589–1600.
- Biginelli, P. Synthesis of 3,4-dihydropyrimidin-2(1H)-ones. Gazz. Chim. Ital. 1893, 23, 360–416.
- Tron, G.C.; Minassi, A.; Appendino, G. Pietro Biginelli: The man behind the reaction. Eur. J. Org. Chem. 2011, 2011, 5541–5550.
- Marinescu, M. Biginelli Reaction mediated synthesis of antimicrobial pyrimidine derivatives and their therapeutic properties. Molecules 2021, 26, 6022.
- Costanzo, P.; Nardi, M.; Oliverio, M. Similarity and competition between Biginelli and Hantzsch reactions: An opportunity for modern medicinal chemistry. Eur. J. Org. Chem. 2020, 2020, 3954–3964.
- Chopda, L.V.; Dave, P.N. Recent advances in homogeneous and heterogeneous catalyst in Biginelli reaction from 2015-19: A concise review. ChemistrySelect 2020, 5, 5552–5572.
- Patil, R.V.; Chavan, J.U.; Dalal, D.S.; Shinde, V.-S.; Beldar, A.G. Biginelli reaction: Polymer supported catalytic approaches. ACS Comb. Sci. 2019, 21, 105–148.
- Shumaila, A.M.A.; Al-Thulaia, A.A.N. Mini-review on the synthesis of Biginelli analogs using greener heterogeneous catalysis: Recent strategies with the support or direct catalyzing of inorganic catalysts. Synth. Commun. 2019, 49, 1613–1632.
- Heravi, M.M.; Moradi, R.; Mohammadkhani, L.; Moradi, B. Current progress in asymmetric Biginelli reaction: An update. Mol. Divers. 2018, 22, 751–767.
- Simurova, N.; Maiboroda, O. Biginelli reaction—An effective method for the synthesis of dihydropyrimidine derivatives. Chem. Het. Comp. 2017, 53, 413–415.
- Nagarajaiah, H.; Mukhopadhyay, A.; Moorthy, J.N. Biginelli reaction: An overview. Tetrahedron Lett. 2016, 57, 5135–5149.
- Heravi, M.M.; Ghavidel, M.; Heidari, B. Microwave-assisted Biginelli reaction: An old reaction, a new perspective. Curr. Org. Synth. 2016, 13, 569–600.
- Heravi, M.M.; Asadi, S.; Lashkariani, B.M. Recent progress in asymmetric Biginelli reaction. Mol. Divers. 2013, 17, 389–407.
- Gong, L.-Z.; Chen, X.-H.; Xu, X.-Y. Asymmetric organocatalytic Biginelli reactions. Chem. Eur. J. 2007, 13, 8920–8926.
- Kappe, C.O. 100 years of the Biginelli didydropyrimide synthesis. Tetrahedron 1993, 32, 6937–6963.
- O’Reilly, B.C.; Atwal, K.S. Synthesis of substituted 1,2,3,4-tetrahydro-6-methyl-2-oxo-5-pyrimidinecarboxyllc acid esters: The Biginelli condensation revisited. Heterocycles 1987, 26, 1185–1188.
- Atwal, K.S.; O’Reilly, B.C.; Gougoutas, J.Z.; Malley, M.F. Synthesis of substituted 1,2,3,4-tetrahydro-6-methyl thioxo-5-pyrimidinecarboxylic acid esters. Heterocycles 1987, 26, 1189–1192.
- Atwal, K.S.; Rovnyak, G.C.; O’Reilly, B.C.; Schwartz, J. Substituted 1,4-dihydropyrimidines. 3. Synthesis of selectively functionalized 2-hetero-1,4-dihydropyrimidines. J. Org. Chem. 1989, 54, 5898–5907.
- Rovnyak, G.C.; Kimball, S.D.; Beyer, B.; Cucinotta, G.; DiMarco, J.D.; Gougoutas, J.; Hedberg, A.; Malley, M.; McCarthy, J.P.; Zhang, R.; et al. Calcium entry blockers and activators: Conformational and structural determinants of dihydropyrimidine calcium channel modulators. J. Med. Chem. 1995, 38, 119–129.
- Chikhale, R.V.; Bhole, R.P.; Khedekar, P.B.; Bhusari, K.P. Synthesis and pharmacological investigation of 3-(substituted-1-phenylethanone)-4-(substituted phenyl)-1, 2, 3,4-tetrahydropyrimidine-5-carboxylates. Eur. J. Med. Chem. 2009, 44, 3645–3653.
- Nilsson, B.; Overman, L. Concise synthesis of guanidine-containing heterocycles using the Biginelli reaction. J. Org. Chem. 2006, 71, 7706–7714.
- Ahmad, M.J.; Hassan, S.F.; Nisa, R.U.; Ayub, K.; Nadeem, M.S.; Nazir, S.; Ansari, F.L.; Qureshi, N.A.; Rashid, U. Synthesis, in vitro potential and computational studies on 2-amino-1, 4-dihydropyrimidines as multitarget antibacterial ligands. Med. Chem. Res. 2016, 25, 1877–1894.
- Chen, Q.; Jiang, L.-L.; Chen, C.-N.; Yang, G.-F. The first example of a regioselective Biginelli-like reaction based on 3-alkylthio-5-amino-1,2,4-triazole. J. Heterocycl. Chem. 2009, 46, 139–148.
- Ryabukhin, S.V.; Plaskon, A.S.; Ostapchuk, E.N.; Volochnyuk, D.M.; Tolmachev, A.A. N-Substituted ureas and thioureas in Biginelli reaction promoted by chlorotrimethylsilane: Convenient synthesis of N1-alkyl-, N1-aryl-, and N1,N3-dialkyl-3,4-dihydropyrimidin-2(1H)-(thi)ones. Synthesis 2007, 3, 417–427.
- Pani, M.S.; Arjun, M.; Sridhar, D.; Srinivas, K.; Raviprasad, T. N-Substituted benzoxazolyl ureas and thioureas in Biginelli reaction promoted by trifluoromethane sulfonic acid: An efficient and convenient synthesis of substituted benzoxazolyl 3,4-dihydropyrimidine (1H)-(thio)-ones. Chin. Chem. Lett. 2009, 20, 909–912.
- Rajanarendar, E.; Reddy, M.N.; Murthy, K.R.; Reddy, K.G.; Raju, S.; Srinivas, M.; Praveen, B.; Rao, M.S. Synthesis, antimicrobial, and mosquito larvicidal activity of 1-aryl-4-methyl-3,6-bis-(5-methylisoxazol-3-yl)-2-thioxo-2,3,6,10b-tetrahydro-1H-pyrimidoquinolin-5-ones. Bioorg. Med. Chem. Lett. 2010, 20, 6052–6055.
- Lillo, V.J.; Mansilla, J.; Saá, J.M. Organocatalysis by networks of cooperative hydrogen bonds: Enantioselective direct Mannich addition to preformed arylideneureas. Angew. Chem. Int. Ed. 2016, 55, 4312–4316.
- Ninomiya, M.; Garud, D.R.; Koketsu, M. Selenium-containing heterocycles using selenoamides, selenoureas, selenazadienes, and isoselenocyanates. Heterocycles 2010, 81, 2027–2055.
- Samdhiana, V.; Bhatiaa, S.K.; Kaura, B. Cell-viability analysis against MCF-7 human breast cell line and antimicrobial evaluation of newly synthesized selenoxopyrimidines. Russ. J. Org. Chem. 2019, 55, 1041–1046.
- Krauskopf, F.; Truong, K.-N.; Rissanen, K.; Bolm, C. 2,3-Dihydro-1,2,6-thiadiazine 1-oxides by Biginelli-type reactions with sulfonimidamides under mechanochemical conditions. Org. Lett. 2021, 23, 2699–2703.
- Khabazzadeh, H.; Saidi, K.; Sheibani, H. Highly efficient conversion of aromatic acylals to 3, 4-dihydropyrimidinones: A new protocol for the Biginelli reaction. Arkivoc 2008, 2008, 34–41.
- Stucchi, M.; Lesma, G.; Meneghetti, F.; Rainoldi, G.; Sacchetti, A.; Silvani, A. Organocatalytic asymmetric Biginelli-like reaction involving isatin. J. Org. Chem. 2016, 81, 1877–1884.
- Malik, A.A.; Dangroo, N.A.; Ara, T. Microwave-assisted tandem Kornblum oxidation and Biginelli reaction for the synthesis of dihydropyrimidones. ChemistrySelect 2020, 5, 12965–12970.
- Kshirsagar, S.W.; Patil, N.R.; Samant, S.D. Mg–Al hydrotalcites as the first heterogeneous basic catalysts for the Kornblum oxidation of benzyl halides to benzaldehydes using DMSO. Tetrahedron Lett. 2008, 49, 1160–1162.
- Shaabani, A.; Bazgir, A.; Teimouri, F. Ammonium chloride-catalyzed one-pot synthesis of 3,4-dihydropyrimidin-2-(1H)-ones under solvent-free conditions. Tetrahedron Lett. 2003, 44, 857–859.
- Darehkordi, A.; Hosseini, M.S. Montmorillonite modified as an efficient and environment friendly catalyst for one- pot synthesis of 3, 4-dihydropyrimidine-2(1H) ones. Iran. J. Mater. Sci. Eng. 2012, 9, 49–57.
- Matias, M.; Campos, G.; Santos, A.O.; Falcão, A.; Silvestre, S.; Alves, G. Potential antitumoral 3,4-dihydropyrimidin-2-(1H)-ones: Synthesis, in vitro biological evaluation and QSAR studies. RSC Adv. 2016, 6, 84943–84958.
- Matias, M.; Campos, G.; Santos, A.O.; Falcão, A.; Silvestre, S.; Alves, G. Synthesis, in vitro evaluation and QSAR modelling of potential antitumoral 3,4-dihydropyrimidin-2-(1H)-thiones. Arab. J. Chem. 2019, 12, 5086–5102.
- Gartner, M.; Sunder-Plassmann, N.; Seiler, J.; Utz, M.; Vernos, I.; Surrey, T.; Giannis, A. Development and biological evaluation of potent and specific inhibitors of mitotic kinesin Eg5. ChemBioChem 2005, 6, 1173–1177.
- Gemal, A.L.; Luche, J.L. Lanthanoids in organic synthesis. 6. Reduction of alpha-enones by sodium borohydride in the presence of lanthanoid chlorides: Synthetic and mechanistic aspects. J. Am. Chem. Soc. 1981, 103, 5454–5459.
- Kidwai, M.; Saxena, S.; Khan, M.K.R.; Thukral, S.S. Synthesis of 4-aryl-7,7-dimethyl-1,2,3,4,5,6,7,8-octahydroquinazoline-2-one/thione-5-one derivatives and evaluation as antibacterials. Eur. J. Med. Chem. 2005, 40, 816–819.
- Abnous, K.; Barati, B.; Mehri, S.; Farimani, M.R.M.; Alibolandi, M.; Mohammadpour, F.; Ghandadi, M.; Hadizadeh, F. Synthesis and molecular modeling of six novel monastrol analogues: Evaluation of cytotoxicity and kinesin inhibitory activity against HeLa cell line. J. Pharm. Sci. 2013, 21, 70.
- Niralwad, K.S.; Shingate, B.B.; Shingare, M.S. Microwave-assisted one-pot synthesis of octahydroquinazolinone derivatives using ammonium metavanadate under solvent-free conditions. Tetrahedron Lett. 2010, 51, 3616–3618.
- Badadhe, P.V.; Chate, A.V.; Hingane, D.G.; Mahajan, P.S.; Chavhan, N.M.; Gill, C.H. Microwave-assisted one-pot synthesis of octahydroquinazolinone derivatives catalyzed by thiamine hydrochloride under solvent-free condition. J. Korean Chem. Soc. 2011, 55, 936–939.
- Shah, P.M.; Patel, M.P. Zn(OTf)2-catalyzed three component, one-pot cyclocondensation reaction of some new octahydroquinazolinone derivatives and access their bio-potential. Med. Chem. Res. 2012, 21, 1188–1198.
- Silva, G.C.O.; Correa, J.R.; Rodrigues, M.O.; Alvim, H.G.O.; Guido, B.C.; Gatto, C.C.; Wanderley, K.A.; Fioramonte, M.; Gozzo, F.C.; de Souza, R.O.M.A.; et al. The Biginelli reaction under batch and continuous flow conditions: Catalysis, mechanism and antitumoral activity. RSC Adv. 2015, 5, 48506–48515.
- Bariwal, J.J.; Malhotra, M.; Molnar, J.; Jain, K.S.; Shah, A.K.; Bariwal, J.B. Synthesis, characterization and anticancer activity of 3-aza-analogues of DP-7. Med. Chem. Res. 2012, 21, 4002–4009.
- Agbaje, O.C.; Fadeyi, O.O.; Fadeyi, S.A.; Myles, L.E.; Okoro, C.O. Synthesis and in vitro cytotoxicity evaluation of some fluorinated hexahydropyrimidine derivatives. Bioorg. Med. Chem. Lett. 2011, 21, 989–992.
- Azizian, J.; Mohammadi, M.K.; Firuzi, O.; Mirza, B.; Miri, R. Microwave-Assisted Solvent-Free Synthesis of Bis(Dihydropyrimidinone)Benzenes and Evaluation of Their Cytotoxic Activity. Chem. Biol. Drug Des. 2010, 75, 375–380.
- Zhu, L.; Cheng, P.; Lei, N.; Yao, J.; Sheng, C.; Zhuang, C.; Guo, W.; Liu, W.; Zhang, Y.; Dong, G.; et al. Synthesis and biological evaluation of novel homocamptothecins conjugating with dihydropyrimidine derivatives as potent topoisomerase I inhibitors. Arch. Pharm. 2011, 344, 726–734.
- Gijsen, H.J.M.; Berthelot, D.; De Cleyn, M.A.J.; Geuens, I.; Brône, B.; Mercken, M. Tricyclic 3,4-dihydropyrimidine-2-thione derivatives as potent TRPA1 antagonists. Bioorg. Med. Chem. Lett. 2012, 22, 797–800.
- Rajitha, G.; Arya, C.G.; Janardhan, B.; Laxmi, S.V.; Ramesh, G.; Kumari, U.S. 3-Aryl/heteryl-5-phenylindenothiazolopyrimidin-6(5H)-ones: Synthesis, characterization, and antimicrobial investigation. Russ. J. Bioorg. Chem. 2020, 46, 612–619.
- Lal, J.; Gupta, S.K.; Thavaselvam, D.; Agarwal, D.D. Design, synthesis, synergistic antimicrobial activity and cytotoxicity of 4-aryl substituted 3,4-dihydropyrimidinones of curcumin. Bioorg. Med. Chem. Lett. 2012, 22, 2872–2876.
- Lal, J.; Gupta, S.K.; Agarwal, D.D. Chitosan: An Efficient Biodegradable and Recyclable Green Catalyst for One-Pot Synthesis of 3,4-Dihydropyrimidinones of Curcumin in Aqueous Media. Catal. Commun. 2012, 27, 38–43.
- Lal, J.; Gupta, S.K.; Thavaselvam, D.; Agarwal, D.D. Synthesis and pharmacological activity evaluation of curcumin derivatives. Chin. Chem. Lett. 2016, 27, 1067–1072.
- Cheng, Q.; Wang, Q.; Tan, T.; Chen, N.; Shuai, M. One-pot synthesis of 13-acetyl-9-methyl-11-oxo(thioxo)-8-oxa-10,12-diazatricyclotrideca-2,4,6-trienes under microwave irradiation. J. Heterocycl. Chem. 2012, 49, 1352–1356.
- Patil, B.M.; Shinde, S.K.; Jagdale, A.A.; Jadhav, S.D.; Patil, S.S. Fruit extract of averrhoa bilimbi: A green neoteric micellar medium for isoxazole and biginelli-like synthesis. Res. Chem. Intermed. 2021, 47, 4369–4398.
- Ramos, L.M.; Guido, B.C.; Nobrega, C.C.; Corrêa, J.R.; Silva, R.G.; de Oliveira, H.C.B.; Gomes, A.F.; Gozzo, F.C.; Neto, B.A.D. The Biginelli reaction with an imidazolium–tagged recyclable iron catalyst: Kinetics, mechanism, and antitumoral activity. Chem. Eur. J. 2013, 19, 4156–4168.
- Guido, B.C.; Ramos, L.M.; Nolasco, D.O.; Nobrega, C.C.; Andrade, B.Y.G.; Pic-Taylor, A.; Neto, B.A.D.; Corrêa, J.R. Impact of kinesin Eg5 inhibition by 3,4-dihydropyrimidin-2(1H)-one derivatives on various breast cancer cell features. BMC Cancer 2015, 15, 283.
- Siddiqui, Z.N.; Khan, T. Unprecedented single-pot protocol for the Synthesis of novel bis-3,4-dihydropyrimidin-2(1H)-ones using PEG-HClO4 as a biodegradable, highly robust and reusable solid acid green catalyst under solvent-free conditions. RSC Adv. 2014, 4, 2526–2537.
- Davanagere, P.M.; Maiti, B. 1,3-Bis(carboxymethyl)imidazolium chloride as a sustainable, recyclable, and metal-free ionic catalyst for the Biginelli multicomponent reaction in neat condition. ACS Omega 2021, 6, 26035–26047.
- El-Emary, T.I.; Abdel-Mohsen, S.A.; Mohamed, S.A. An efficient synthesis and reactions of 5-acetyl-6-methyl-4-(1,3-diphenyl-1H-pyrazol-4-yl)-3,4-dihydropyrmidin-2(1H)-thione as potential antimicrobial and anti-inflammatory agents. Russ. J. Bioorg. Chem. 2021, 47, 561–571.
- Shaabani, A.; Bazgir, A. Microwave-assisted efficient synthesis of spiro-fused heterocycles under solvent-free conditions. Tetrahedron Lett. 2004, 45, 2575–2577.
- Shaabani, A.; Bazgir, A.; Bijanzadeh, H.R. A reexamination of Biginelli-like multicomponent condensation reaction: One-pot regioselective synthesis of spiro heterobicyclic rings. Mol. Divers. 2004, 8, 141–145.
- Mohammadi, G.; Faramarzi, S.; Asadi, S.; Amanlou, M. Green synthesis and urease inhibitory activity of spiro-pyrimidinethiones/spiro-pyrimidinones-barbituric acid derivatives. Iran. J. Pharm. Res. 2015, 14, 1105–1114.
- Dabholkar, V.V.; Tripathi, R.D. Synthesis of Biginelli products of thiobarbituric acids and their antimicrobial activity. J. Serb. Chem. Soc. 2010, 75, 1033–1040.
- Gupta, R.; Jain, A.; Joshi, R.; Jain, M. Eco-friendly solventless synthesis of 5-indolylpyrimidopyrimidinones and their antimicrobial activity. Bull. Korean Chem. Soc. 2011, 32, 899–904.
- Rimaz, M.; Khalafy, J.; Mousavi, H. A Green organocatalyzed one-pot protocol for efficient synthesis of new substituted pyrimidopyrimidinones using a Biginelli-like reaction. Res. Chem. Intermed. 2016, 42, 8185–8200.
- Rimaz, M.; Khalafy, J.; Mousavi, H.; Bohlooli, S.; Khalili, B. Two different green catalytic systems for one-pot regioselective and chemoselective synthesis of some pyrimidopyrimidinone derivatives in water. J. Heterocycl. Chem. 2017, 54, 3174–3186.
- Kamal, A.; Malik, M.S.; Bajee, S.; Azeeza, S.; Faazil, S.; Ramakrishna, S.; Naidu, V.G.M.; Vishnuwardhan, M.V.P.S. Synthesis and biological evaluation of conformationally flexible as well as restricted dimers of monastrol and related dihydropyrimidones. Eur. J. Med. Chem. 2011, 46, 3274–3281.
- Yadlapalli, R.K.; Chourasia, O.P.; Vemuri, K.; Sritharan, M.; Perali, R.S. Synthesis and in vitro anticancer and antitubercular activity of diarylpyrazole ligated dihydropyrimidines possessing lipophilic carbamoyl group. Bioorg. Med. Chem. Lett. 2012, 22, 2708–2711.
- Elumalai, K.; Ali, M.A.; Elumalai, M.; Eluri, K.; Srinivasan, S. Design, synthesis and antimycobacterial activity of some novel 3,5-dichloro-2-ethoxy-6-fluoropyridin-4-amine cyclocondensed dihydropyrimidines. J. Pharm. Res. 2013, 7, 241–245.
- Elumalai, K.; Ali, M.A.; Elumalai, M.; Eluri, K.; Srinivasan, S.; Mohanty, S.K. Microwave-assisted synthesis of some novel acetazolamide cyclocondensed 1,2,3,4-tetrahydropyrimidines as a potent antimicrobial and cytotoxic agents. Beni-Suef Univ. J. Basic Appl. Sci. 2014, 3, 24–31.
- Elumalai, K.; Ali, M.A.; Srinivasan, S.; Elumalai, M.; Eluri, K. Antimicrobial and in vitro cytotoxicity of novel sulphanilamide condensed 1,2,3,4-tetrahydropyrimidines. J. Taibah Univ. Sci. 2017, 11, 46–56.
- Chikhale, R.; Menghani, S.; Babu, R.; Bansode, R.; Bhargavi, G.; Karodia, N.; Rajasekharan, M.V.; Paradkar, A.; Khedekar, P. Development of selective DprE1 inhibitors: Design, synthesis, crystal structure and antitubercular activity of benzothiazolylpyrimidine-5-carboxamides. Eur. J. Med. Chem. 2015, 96, 30–46.
- Ramachandran, V.; Arumugasamy, K.; Singh, S.K.; Edayadulla, N.; Ramesh, P.; Kamaraj, S.-K. Synthesis, antibacterial studies, and molecular modeling studies of 3,4-dihydropyrimidinone compounds. J. Chem. Biol. 2016, 9, 31–40.
- Gein, V.L.; Zamaraeva, T.M.; Fedotov, A.Y.; Balandina, A.V.; Dmitriev, M.V. Synthesis, structure, and antimicrobial activity of N,6-diaryl-4-methyl-2-oxo-1,2,3,6-tetrahydropyrimidine-5-carboxamides. Russ. J. Gen. Chem. 2016, 86, 2437–2441.
- Faizan, S.; Kumar, B.P.; Naishima, N.L.; Ashok, T.; Justin, A.; Kumar, M.V.; Chandrashekarappa, R.B.; Raghavendra, N.M.; Kabadi, P.; Adhikary, L. Design, parallel synthesis of Biginelli 1,4-dihydropyrimidines using PTSA as a catalyst, evaluation of anticancer activity and structure activity relationships via 3D QSAR Studies. Bioorg. Chem. 2021, 117, 105462.
- Bussolari, J.C.; McDonnell, P.A. A new substrate for the Biginelli cyclocondensation: Direct preparation of 5-unsubstituted 3,4-dihydropyrimidin-2(1H)-ones from a β-Keto carboxylic acid. J. Org. Chem. 2000, 65, 6777–6779.
- Fang, Z.; Lam, Y. A rapid and convenient synthesis of 5-unsubstituted 3,4-dihydropyrimidin-2-ones and thiones. Tetrahedron 2011, 67, 1294–1297.
- Bahekar, S.S.; Shinde, D.B. Synthesis and anti-inflammatory activity of some -acetic acid derivatives. Bioorg. Med. Chem. Lett. 2004, 14, 1733–1736.
- Mokale, S.N.; Shinde, S.S.; Elgire, R.D.; Sangshetti, J.N.; Shinde, D.B. Synthesis and anti-inflammatory activity of some 3-(4,6-disubtituted-2-thioxo-1,2,3,4-tetrahydropyrimidin-5-yl) propanoic acid derivatives. Bioorg. Med. Chem. Lett. 2010, 20, 4424–4426.
- Li, N.; Chen, X.-H.; Song, J.; Luo, S.-W.; Fan, W.; Gong, L.-Z. Highly enantioselective organocatalytic Biginelli and Biginelli-like condensations: Reversal of the stereochemistry by tuning the 3,3′-disubstituents of phosphoric acids. J. Am. Chem. Soc. 2009, 131, 15301–15310.
- Holla, B.S.; Rao, B.S.; Sarojini, B.; Akberali, P. One pot synthesis of thiazolodihydropyrimidinones and evaluation of their anticancer activity. Eur. J. Med. Chem. 2004, 39, 777–783.
- Liang, B.; Wang, X.; Wanga, J.-X.; Du, Z. New three-component cyclocondensation reaction: Microwaveassisted one-pot synthesis of 5-unsubstituted-3,4-dihydropyrimidin-2(1H)-ones under solvent-free conditions. Tetrahedron 2007, 63, 1981–1986.
- Safari, J.; Gandomi-Ravandi, S. MnO2–MWCNT nanocomposites as efficient catalyst in the synthesis of Biginelli-type compounds under microwave radiation. J. Mol. Catal. A Chem. 2013, 373, 72–77.
- Desai, N.C.; Joshi, S.B.; Jadeja, K.A. A one-pot multicomponent Biginelli reaction for the preparation of novel pyrimidinthione derivatives as antimicrobial agents. J. Heterocycl. Chem. 2020, 57, 791–795.
- Barbero, M.; Cadamuro, S.; Dughera, S. A Brønsted acid catalysed enantioselective Biginelli reaction. Green Chem. 2017, 19, 1529–1535.
- Hu, X.-Y.; Guo, J.-X.; Wang, C.; Zhang, R.; Borovkov, V. Stereoselective Biginelli-like reaction catalysed by a chiral phosphoric acid bearing two hydroxy groups. Beilstein J. Org. Chem. 2020, 16, 1875–1880.
- Zhu, Y.-L.; Huang, S.-L.; Pan, Y.-J. Highly Chemoselective multicomponent Biginelli-type condensations of cycloalkanones, urea or thiourea and aldehydes. Eur. J. Org. Chem. 2005, 2005, 2354–2367.
- Rahman, M.; Majee, A.; Hajra, A. Microwave-assisted Brønsted acidic ionic liquid-promoted one-pot synthesis of heterobicyclic dihydropyrimidinones by a three-component coupling of cyclopentanone, aldehydes, and urea. J. Heterocycl. Chem. 2010, 47, 1230–1233.
- Zhou, Z.-L.; Wang, P.-C.; Lu, M. Bronsted acidic ionic liquid HSO4 catalyzed one-pot three-component Biginelli-type reaction: An efficient and solvent-free synthesis of pyrimidinone derivatives and its mechanistic study. Chin. Chem. Lett. 2016, 27, 226–230.
- Gogoi, P.; Dutta, A.K.; Borah, R. Studies on structural changes and catalytic activity of Y-zeolite composites of 1,3-disulfoimidazolium trifluoroacetate ionic liquid (IL) for three-component synthesis of 3,4-dihydropyrimidinones. Catal. Lett. 2017, 147, 674–685.
- Amoozadeh, A.; Rahmani, S.; Nemati, F. Poly(ethylene)glycol/AlCl3 as a new and efficient system for multicomponent Biginelli-type synthesis of pyrimidinone derivatives. Heterocycl. Commun. 2013, 19, 69–73.
- Kaur, R.; Bansal, M.; Kaur, B.; Mishra, T.; Bhatia, A. Synthesis of 4-aryl-4,5-dihydro-1H-indenopyrimidines by Biginelli condensation and their antibacterial activities. J. Chem. Sci. 2011, 123, 443–451.
- Velpula, R.; Banothu, J.; Gali, R.; Deshineni, R.; Bavantula, R. 1-Sulfopyridinium chloride: Green and expeditious ionic liquid for the one-pot synthesis of fused 3,4-dihydropyrimidin-2(1H)-ones and thiones under solvent-free conditions. Chin. Chem. Lett. 2015, 26, 309–312.
- Qu, H.; Li, X.; Mo, F.; Lin, X. Efficient synthesis of dihydropyrimidinones via a three-component Biginelli-type reaction of urea, alkylaldehyde and arylaldehyde. Beilstein J. Org. Chem. 2013, 9, 2846–2851.
- Bhat, M.A.; Al-Dhfyan, A.; Al-Omar, M.A. Targeting cancer stem cells with novel 4-(4-substituted phenyl)-5-(3,4,5-trimethoxy/3,4-dimethoxy)-benzoyl-3,4-dihydropyrimidine-2(1H)-one/thiones. Molecules. 2016, 21, 1746.
- Carreiro, E.P.; Sena, A.M.; Puerta, A.; Padrón, J.M.; Burke, A.J. Synthesis of novel 1,2,3-triazole-dihydropyrimidinone hybrids using multicomponent 1,3-dipolar cycloaddition (click)–Biginelli reactions: Anticancer activity. Synlett 2020, 31, 615–621.
- Mohammed, S.M.; Moustafa, A.H.; Ahmed, N.; El-Sayed, H.A.; Mohamed, A.S.A. Nano-K2CO3-catalyzed Biginelli-type reaction: Regioselective synthesis, DFT study, and antimicrobial activity of 4-aryl-6-methyl-5-phenyl-3,4-dihydropyrimidine-2(1H)-thiones. Russ. J. Org. Chem. 2022, 58, 136–143.

