The interest in 3,4-dihydropyrimidine-2(1H)-(thio)ones is increasing every day, mainly due to their paramount biological relevance. The Biginelli reaction is the classical approach to reaching these scaffolds, although the product diversity suffers from some limitations. In order to overcome these restrictions, two main approaches have been devised. The first one involves the modification of the conventional components of the Biginelli reaction and the second one refers to the postmodification of the Biginelli products. Both strategies have been extensively revised in this manuscript. Regarding the first one, initially, the modification of one of the components was covered. Although examples of modifications of the three of them were described, by far the modification of the keto ester counterpart was the most popular approach, and a wide variety of different enolizable carbonylic compounds were used; moreover, changes in two or the three components were also described, broadening the substitution of the final dihydropyrimidines. Together with these modifications, the use of Biginelli adducts as a starting point for further modification was also a very useful strategy to decorate the final heterocyclic structure.
1. Introduction
The recurrent presence of some structural fragments in biologically important compounds and drugs with diverse biological activities was used by Evans to introduce the term “privileged structures” in 1988
[1] (later updated by Patchett and Nargund)
[2]; these scaffolds are able to interact with more than one receptor or enzyme, playing a relevant role as a starting point in the drug discovery process. Among the fragments labelled as “privileged structures”, 3,4-dihydropyrimidinones (DHPMs) and their derivatives occupy a prominent place; these cores are of immense biological importance; play an important role as essential building blocks in the synthesis of DNA and RNA, and subtle changes in their structure provide a wide range of biological activities such as anti-inflammatory, anti-HIV, anti-tubercular, antifungal, anticancer, antibacterial, antifilarial, antihyperglycemic, antihypertensive, analgesic, anticonvulsant, antioxidant, anti-TRPA1 or anti-SARS, among others
[3][4][5][6][7][8][9][10][3,4,5,6,7,8,9,10]; it is not surprising that the popularity of DHPMs lasts until present days, and the development of new methodologies to access these structural motifs is always of high interest
[11][12][13][14][11,12,13,14].
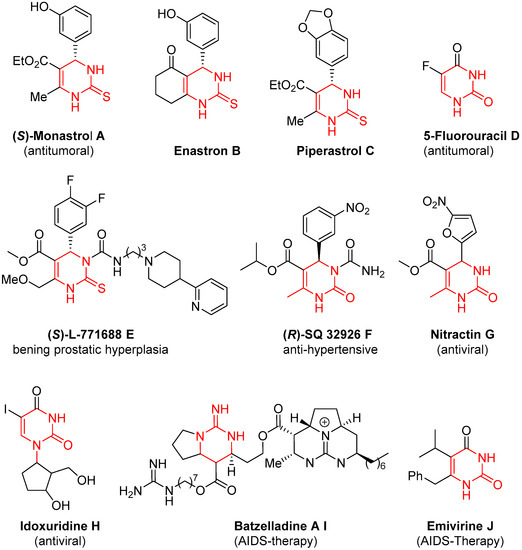
Emblematic examples of DHPM-derivatives are monastrol and its derivatives enastron and piperastrol
[15]; these compounds inhibit Kinesin-5, a protein involved in the regulation and function of mitosis, and are considered promising targets in cancer chemotherapy (
Figure 1A–C). 5-Fluorouracil
[16], due to its analogy with uracil, inhibits DNA formation by irreversible union with thimidilate synthase enzyme, inducing cell death (
Figure 1D). After the discovery of the antitumoral properties of 5-fluorouracil, the incorporation of fluorinated moieties into organic molecules became a fundamental strategy in medicinal chemistry. (
S)-L-771688 is the first α
1a-adrenoceptor selective antagonist to be tested in the clinic for the treatment of benign prostatic hyperplasia (
Figure 1E)
[17]. (
R)-SQ 32926, considered a close structural analog of the therapeutically widely used calcium channel blockers of the 1,4-dihydropyridine type (e.g., nifedipine) displayed interesting hypertensive properties (
Figure 1F)
[18]. Nitractin is highly effective against trachoma virus and also shows some antibacterial activity (
Figure 1G)
[19]. Idoxuridine, initially developed as an anticancer drug, became an antiviral agent used for the topical treatment of herpes simplex keratitis (
Figure 1H)
[20]. Batzelladine A belongs to a family of polycyclic guanidine alkaloids that inhibit the binding of HIVgp-120-CD4 (
Figure 1I)
[21]. Finally, emivirine was developed as an agent for the treatment of HIV as a non-nucleoside reverse transcriptase inhibitor (
Figure 1J)
[22].
Figure 1.
Representative examples (
A
–
J
) of biologically active DHPM-derivatives.
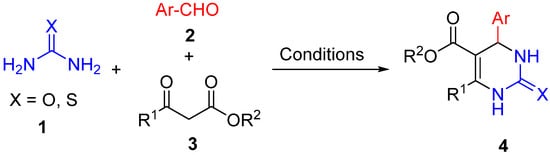
The classical approach to accessing DHPM derivatives is the Biginelli reaction. Initially, it involved the acid-catalyzed cyclocondensation of a urea or thiourea
1, aromatic aldehyde
2, and a beta-keto ester
3 (
Scheme 1). First described in 1893
[23][24][23,24], it remained almost unexplored until the 1980s when the power of multicomponent reactions was recognized as a useful tool for medicinal chemists. The convergent character, operational simplicity, easily accessible and molecular diverse starting materials as well as the atom economy of this transformation make the Biginelli reaction one of the most important multicomponent processes in drug discovery, that continuously attracts research interests due to the occurrence of DHPMs in biologically active products and drugs. On account of this research activity, a wide variety of methodologies have been devised for the racemic and asymmetric Biginelli reaction, together with a big number of reaction conditions that involved homogeneous and heterogeneous catalysis, the use of ionic liquids as the solvent, immobilized catalysts in solid supports or the use of microwave irradiation
[25][26][27][28][29][30][31][32][33][34][35][36][25,26,27,28,29,30,31,32,33,34,35,36]. Additionally, a big number of variants of this multicomponent reaction that gain access to novel DHPMs by modification of the original fragments in a multicomponent or step-wise manner have been described.
Scheme 1.
Classical Biginelli reaction.
2. Modification of the Urea Counterpart
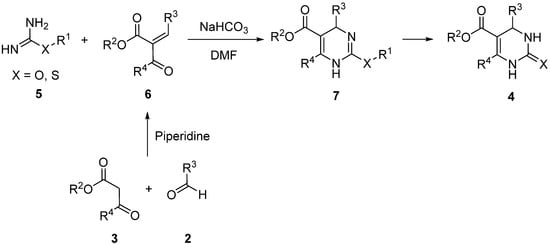
The first important change of the urea counterpart in a Biginelli-type reaction was the Atwal modification
[37][38][39][37,38,39]. In this approach,
O,
S-substituted isoureas
5 together with a preformed unsaturated carbonyl compound
6 were condensed in a basic medium (
Scheme 2). Through this methodology, it was possible to improve the efficiency of the Biginelli synthesis, especially with aliphatic and aromatic aldehydes slightly hindered by
ortho-substituents. The unsaturated carbonyl compound
6 was obtained via Knoevenagel condensation from the corresponding β-keto esters
3 and aldehydes
2 in a separated synthesis.
Scheme 2.
Atwal modification of the Biginelli synthesis.
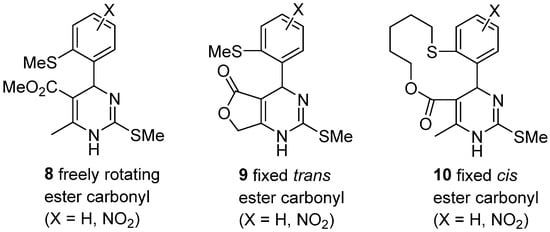
Using this approach, Rovnyak et al. prepared uniquely designed dihydropyrimidines
8–
10 starting from conveniently functionalized α-benzylidene β-keto esters
6; these derivatives were synthesized in order to establish structural and conformational determinants in calcium channel modulation (
Scheme 3)
[40].
Scheme 3.
Structural and conformational restrictions of calcium channel modulators 8
–10
.
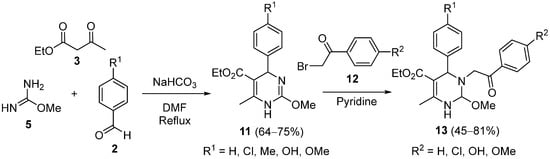
However, preformation of the α-benzylidene β-keto esters was not mandatory in all cases and the classical three-component reaction could be performed directly in a basic medium using
O-methyl isourea
5, ethyl acetoacetate
3 and substituted benzaldehydes
2 to form DHPMs
11 (
Scheme 4); these compounds were derivatized through the selective reaction with phenacyl bromides
12 at N3, to obtain derivatives
13 that showed good antihypertensive, anti-inflammatory, and analgesic activity, as well as low ulcerogenic activity. The N3 selectivity could be a consequence of the richer electron density of N3 compared to N1
[41].
Scheme 4.
Synthesis of DHPMs derivatives 13
with antihypertensive, anti-inflammatory and analgesic activity.
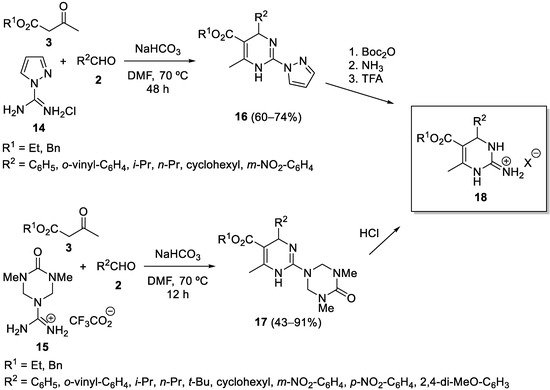
Some other variations of the urea component in Biginelli-like reactions include the use of guanidine to obtain 2-amino-1,4-dihydropyrimidines, however, the direct three-component Biginelli reaction with guanidine is useful only with benzoyl acetates and aryl aldehydes, and fails to give useful yields using acetoacetates. A more general Biginelli-based method for preparing 2-imino-5-carboxy-3,4-dihydropyrimidines
18 was developed by Nilsson and Overman in 2006
[42]. Two alternatives are shown in
Scheme 5, starting from pyrazole carboxamidine
14 in a four-step sequence, or starting from the triazone-protected guanidine
15 in a two-step sequence. Both alternatives utilize acetoacetates
3 and are compatible with aliphatic aldehydes.
Scheme 5.
Synthesis of 2-imino-5-carboxy-3,4-dihydropyrimidines 18
.
Ultrasound irradiation has also been used to promote the direct three-component Biginelli reaction with guanidine hydrochloride, acetoacetates and aromatic aldehydes
[43].
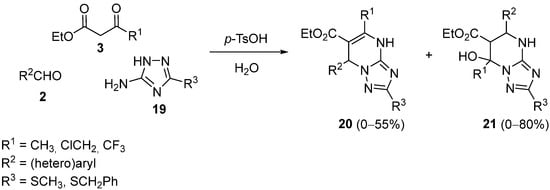
At the beginning of the XXI century, the Biginelli reaction was extended by replacing the urea component with 5-amino-1,2,4-triazoles. Theoretically, four possible compounds could be obtained taking into account two regioisomers with or without dehydration. In fact, 3-alkylthio-5-amino-1,2,4-triazoles
19 gave two different dihydrotriazolo-pyrimidines
20 and
21 (
Scheme 6), the selectivity strongly depending on the substitution of the reactants
[44].
Scheme 6.
Synthesis of dihydrotriazolo-pyrimidine pseudo
-regioisomers 20
and 21
.
N-Substituted ureas and thioureas have also been used in Biginelli-like reactions
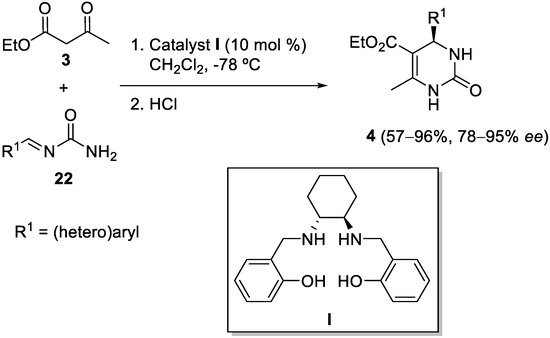
[45][46][47][45,46,47]. Inspired by the mechanisms of biocatalysts, in 2016, the Saá research group developed the concept of noncovalent organocatalysis by means of networks of cooperative hydrogen bonds, utilizing arylideneureas
22 in the asymmetric reaction with ethyl acetoacetate
3 (
Scheme 7). Arylideneureas can act as donor-acceptor in hydrogen bonds, thus they are capable of assembling with the chiral catalyst
I and at the same time activate the nucleophile, rendering final DHPMs
4 with excellent enantioselectivities
[48].
Scheme 7.
Arylideneureas 22
as substrates in the asymmetric synthesis of DHPMs 4
.
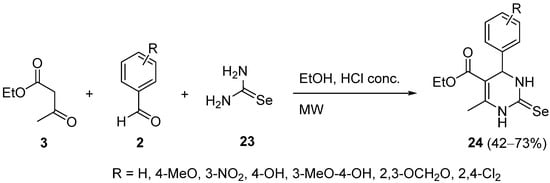
The use of selenourea
23 as the starting material is one of the most efficient methods for the synthesis of selenium-containing heterocycles
[49], and it has been used to prepare selenoxopyrimidines
24 by means of a one-pot multicomponent reaction with ethyl acetoacetate
3 and aromatic aldehydes
2 in acidic medium (
Scheme 8). The synthesized compounds were shown to possess a significant antimicrobial and anticancer activity in vitro
[50].
Scheme 8.
Synthesis of selenoxopyrimidines 24
.
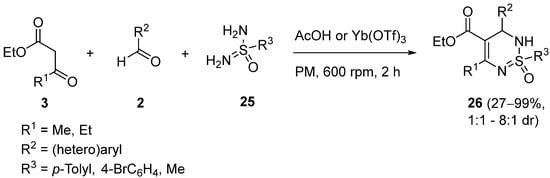
Finally,
NH-free sulfonimidamides
25 have also been used as the urea component in Biginelli-type multicomponent reactions (MCRs), to provide 2,3-dihydro-1,2,6-thiadiazine 1-oxides
26, generally in high yields (
Scheme 9). As the sulphur in sulfonimidamides is stereogenic the reaction produces two diastereoisomers with variable selectivities. The couplings are performed in a planetary ball mill (PM) under solvent-free mechanochemical conditions, catalysed by acetic acid or ytterbium triflate
[51].
Scheme 9.
Synthesis of 2,3-dihydro-1,2,6-thiadiazine 1-oxides 26
.
3. Modification of the Aldehyde Counterpart
The original Biginelli reaction was described with aromatic aldehydes. Although the reaction with aliphatic aldehydes is less efficient, their participation in this MCR is now widespread and, in this section, the use of aliphatic aldehydes will not be considered as a modification of the aldehyde component.
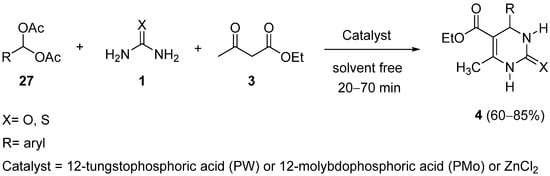
The first reported example of a modification at the aldehyde building block in the Biginelli reaction was the use of acylals
27 [52]; this masked carbonyl functionality, together with ethyl acetoacetate
3 and urea or thiourea
1 were employed to give dihydropyrimidinones
4 under acid catalysis. The reaction was catalyzed by 12-tungstophosphoric acid (PW), 12-molybdophosphoric acid (PMo) or zinc chloride and performed in a one-pot procedure under solvent-free conditions (
Scheme 10).
Scheme 10.
Synthesis of DHPMs 4
from acylals 27
as the aldehyde component.
The best conditions to prepare the DHPMs were achieved when 10 mol%, 20 mol%, and 80 mol% of PW, PMo, and ZnCl2 were used, respectively; these optimum conditions were applied to a series of substituted aromatic acylals 27 observing that with electron-donating containing substituents the reaction proceeded faster and gave high yields in short reaction times.
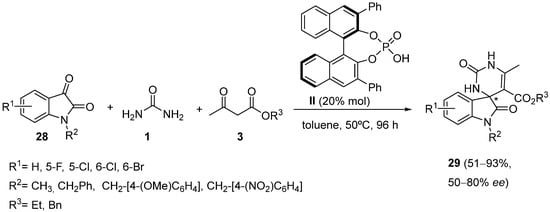
Stucchi et al. described the use of
N-substituted isatins
28 as carbonyl substrates in an asymmetric, Brønsted acid catalyzed Biginelli-like reaction
[53]. The use of BINOL-derived phosphoric acid catalyst
II allowed the authors to obtain enantioenriched spiro(indoline-pyrimidine)-diones derivatives
29 in moderate-to-good yields (
Scheme 11).
Scheme 11.
Synthesis of spiro(indoline-pyrimidine)-diones 29
.
The halogen substitution at the aryl ring had little effect on both yield and ee. Likewise, methyl and benzyl acetoacetates provided similar results leading to good yields and moderate ee’s in the final products; however, the N-Me isatin gave a better result than the corresponding N-benzyl, N-p-nitrobenzyl, and N-p-methoxybenzyl ones in terms of yield (93% to up to 63%), although suffering a drop in ee (50% to up to 80%). Surprisingly, neither thiourea in place of urea nor various linear or cyclic β-diketones instead of alkyl acetoacetates gave good results. With thiourea, no reaction occurred, whereas, with β-diketones, a complex mixture of products was obtained.
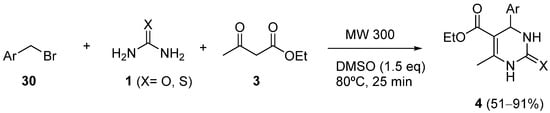
Recently, A. A. Malik and coworkers described the synthesis of dihydropyrimidones
4 via sequential Kornblum oxidation/Biginelli reaction
[54]. The method involves the in-situ generation of benzaldehydes
2 from benzyl halides
30, under catalyst-free conditions, which were subsequently converted into dihydropyrimidones
4 in a one-pot manner under microwave (MW) irradiation (
Scheme 12).
Scheme 12.
Synthesis of DHPMs 4
from benzyl bromides 30
as the aldehyde component.
The key step for this transformation was the oxidation of benzyl halide to aldehyde using Kornblum oxidation conditions
[55]. The best synthetic results for this oxidation were obtained when the reaction was performed under microwave irradiation at 80 °C using DMSO as the solvent and in the absence of a catalyst. Under these conditions, the tandem one-pot synthesis of dihydropyrimidones was achieved by reacting benzyl bromide (1.0 mmol) in the presence of urea (1.0 mmol) and ethyl acetoacetate (1.2 mmol) in 1.5 mmol of DMSO.
The scope of the reaction was evaluated using different substituted benzyl halides. In general, both electron-withdrawing and electron-releasing substituents attached to the benzyl substrate reacted successfully and afforded the desired products in good yields; however, benzyl substrates with electron-withdrawing substituents needed more time for completion with lower yields compared to benzyl halides with electron-releasing groups.
The product purification through aqueous recrystallization avoids the use of large quantities of volatile and toxic organic solvents, which makes the method environmentally and nature-friendly.
4. Modification of the Keto Ester Counterpart
4.1. Diketones as the Keto Ester Component
1,3-diketone compounds have been extensively used as substrates for the Biginelli reaction as the enolizable component. RWesearchers will review below the most representative examples.
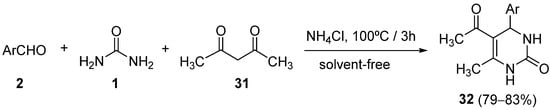
Shaabani and coworkers described the use of ammonium chloride as a catalyst in a one-pot Biginelli condensation reaction of aldehydes
2, 1,3-dicarbonyl compounds
31, and urea or thiourea
1 under solvent-free conditions
[56]. The best results were obtained with a 0.5:1:1:1.5 ratio of ammonium chloride, aldehyde, 1,3-dicarbonyl compound, and urea or thiourea. Particularly, the use of acetylacetone as the dicarbonyl compound afforded the corresponding 3,4-dihydropyrimidin-2-(1
H)-ones
32 with good yields, under these conditions (
Scheme 13).
Scheme 13.
Synthesis of DHPMs 32
from dicarbonyls 31
as ketoester component.
Montmorillonite has also been used as an efficient environmentally friendly catalyst for the synthesis of 3,4-dihydropirimidine-2(1
H)-ones under one-pot three-component Biginelli reaction under solvent-free conditions. The use of acetylacetone as the 1,3-dicarbonyl counterpart afforded the corresponding product in good yield
[57].
More recently, the use of acetylacetone as substrate in a one-pot synthesis of 3,4-dihydropyrimidin-2-(1
H)-ones and 3,4-dihydropyrimidin-2-(1
H)-thiones catalyzed by Bi(NO
3)
3·5H
2O or ZrCl
4, respectively, under solvent-free conditions, was reported by Matias et al.
[58][59][58,59]. The in vitro antiproliferative activity and QSAR studies of the synthesized compounds were also described.
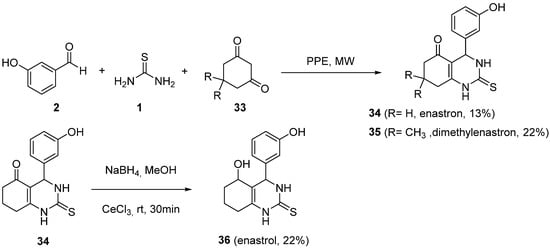
Gartner et al. published the synthesis and biological evaluation of several analogs of monastrol. The different analogs were synthesized as racemic mixtures by using the Biginelli reaction
[60]. The most potent analogs, with enhanced inhibition of Mitotic Kinesin Eg5 compared to monastrol, were those named by the authors as enastron
34, dimethylenastron
35, and enastrol
36 (
Scheme 14). The irradiation of a mixture of a cyclic diketone
33, 3-hydroxybenzaldehyde, and thiourea, in a domestic microwave oven, together with the use of polyphosphate ester (PPE) afforded enastron
34 and dimethylenastron
35 with moderate yields. Enastrol
36 was synthesized as a 3:1 diastereomeric mixture from enastron
34 by selective Luche reduction
[61] of the 5-carbonyl function (
Scheme 14).
Scheme 14.
Synthesis of enastrom 34
, dimethylenastrom 35
and enastrol 36
.
The most potent compound, dimethylenastron 35, is up to more than 100-times more potent than monastrol, both in vitro and with arresting mitosis of cultured cells; these novel inhibitors have the potential to be interesting anticancer drug candidates.
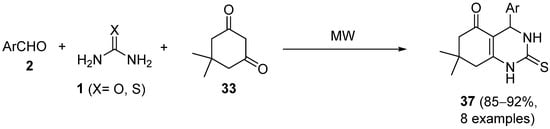
Independently, Kidwai and coworkers described the synthesis of 4-aryl-7,7-dimethyl-1,2,3,4,5,6,7,8-octahydroquinazoline-2-one/thione-5-one derivatives
37 using a Biginelli reaction in the absence of solvent and catalyst and under microwave irradiation, employing neat reaction conditions (
Scheme 15)
[62]. The use of aromatic aldehydes
2, urea/thiourea
1, and 5,5-dimethyl- 1,3-cyclohexanedione
33 (dimedone) as the 1,3-dicarbonyl compound, led to the corresponding quinazoline derivatives
37 in good yields. The synthesized compounds were screened for their in vitro antibacterial activity against standard strains of
Staphylococcus aureus,
Escherichia coli and
Pseudomonas aeruginosa.
Scheme 15.
Synthesis of DHPMs 37
from cyclic diketone 33
as the keto ester component.
Later, Abnous and coworkers reported the synthesis of six Biginelli compounds through one-step Biginelli reaction of dimedone
33 with three imidazole aldehydes, and urea or thiourea using chlorotrimethylsilane (TMSCl) as a catalyst. The products were evaluated for their in vitro cytotoxicities and their inhibitory effects on ATPase activity of kinesin
[63].
Following a similar process, Niralwad et al. described the microwave-assisted one-pot synthesis of octahydroquinazolinone derivatives in high yields using dimedone, urea/thiourea, and appropriate aromatic aldehydes under ammonium metavanadate (NH
4VO
3) as a catalyst under solvent-free conditions
[64]. Likewise, Badadhe and coworkers reported the use of 10 mol% of thiamine hydrochloride (VB1) as an efficient catalyst affording good to excellent yields
[65]. Additionally, Shah and coworkers published the synthesis of some new octahydro-quinazolinone derivatives using zinc triflate (Zn(OTf)
2) as a catalyst, in refluxing ethanol, in high yield
[66].
More recently, 1,3-cyclohexanedione or dimedone has been employed by Silva and coworkers as substrates for the Biginelli synthesis of 3,4-dihydropyrimidin-2(1
H)-one or thione (DHPMs) derivatives catalyzed by two novel coordination polymers (CPs) under solvent-free conditions and heterogeneous catalysis. The reaction conducted under continuous flow conditions afforded very promising results toward a scale-up of the reaction
[67].
Bariwald et al. reported the use of benzoylacetone as the 1,3-dicarbonyl counterpart in the Biginelli reaction together with urea/thiourea and several substituted benzaldehydes in ethanol with a catalytic amount of conc. HCl. The newly synthesized molecules were screened for their anti-proliferative activity
[68].
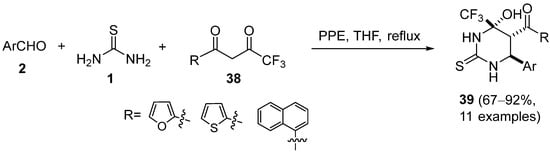
Another example of a Biginelli reaction with PPE catalysis employing 1,3-diketones is the synthesis of a series of trifluoromethylated hexahydropyrimidine and tetrahydropyrimidine derivatives
39 was described by Agbaje et al. (
Scheme 16)
[69]; these fluorinated compounds were evaluated for their in vitro cytotoxic activities in a colon cancer cell line (COLO 320 HSR).
Scheme 16.
Synthesis of trifluoromethylated DHPMs 39
.
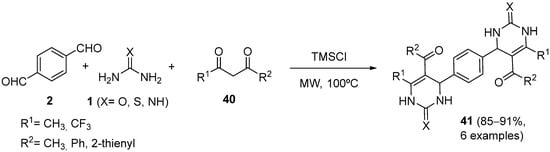
Interestingly, Azizian and coworkers presented the first synthesis of novel derivatives of bis(dihydropyrimidinone)benzenes
41 using chlorotrimethylsilane (TMSCl) as the catalyst through the reaction of terephthalic aldehyde, 1,3-dicarbonyl compounds
40 and (thio)urea or guanidine
1 under microwave irradiation conditions (
Scheme 17)
[70]; this Biginelli condensation method provided products containing two different dihydropyrimidinone units and allowed their obtaining in with high yields (>85%) and in short reaction times (4–6 min).
Scheme 17.
Synthesis of bis-DHPMs 41
.
The cytotoxic activities of these compounds were evaluated on five different human cancer cell lines. Their cytotoxic study indicated that they possessed a weak-to-moderate activity.
The same Biginelli reaction, catalyzed by TMSCl, in dimethylformamide as the solvent, at room temperature, without the use of microwave irradiation, was employed by Zhu et al.
[71], for the synthesis of several dihydropyrimidine derivatives. Three of them derived from the use of 1,3-diketones as the 1,3-dicarbonyl counterpart in the Biginelli reaction. The dihydropyrimidine derivatives were subsequently coupled with homocamptothecin to obtain novel conjugates (hCPT-DHPM); these conjugates were effective cytotoxic agents that showed also superior Topo I inhibition activity than hCPT itself.
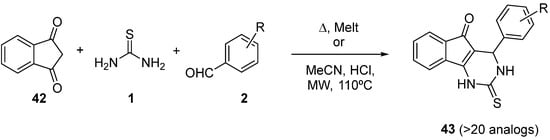
The synthesis of tricyclic 3,4-dihydropyrimidine-2-thione derivatives
43 was described by Gijsen et al.
[72], via a Biginelli three-component reaction between indane-1,3-dione
42, thiourea, and several substituted benzaldehydes
2 (
Scheme 18). Subsequent derivatization led also to some
N-methylated compounds.
Scheme 18.
Synthesis of DHPMs 43
from indane-1,3-dione as ketoester component.
All products were tested on both the human and rat TRPA1 channel. For two of the most interesting compounds, the racemates were separated into the enantiomers, showing that only the dextrorotary enantiomers were active. The absolute configuration of the active enantiomer was determined to be 4R.
Recently, the use of indane-1,3-dione has also been described for the synthesis of 3-substituted 5-phenylindeno-thiazolopyrimidinone derivatives. The Biginelli reaction proceeded under solvent-free conditions, using Poly(4-vinylpyridinium)hydrogen sulfate as the catalyst
[73]. All the synthesized molecules were investigated for their antimicrobial potency against different microbes.
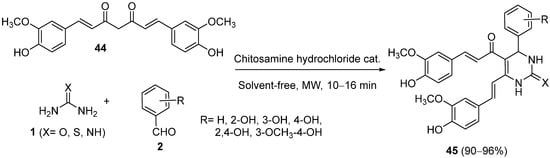
Lal and coworkers described the synthesis of curcumin derivatives using a one-pot cyclocondensation of curcumin (
44) with substituted aromatic aldehydes
2 and urea/thiourea/guanidine
1 in the presence of different catalysts
[74][75][76][74,75,76]. The use of chitosamine hydrochloride as a biodegradable and nontoxic catalyst, under solvent-free conditions and using microwave irradiation, allowed the authors to synthesize curcumin 3,4-dihydropyrimidinones/thiones/imines
45 in excellent yields. All compounds were evaluated for their antioxidant and anti-inflammatory activity (
Scheme 19)
[76].
Scheme 19.
Synthesis of DHPMs 45
from curcumin (44
) as the keto ester component.
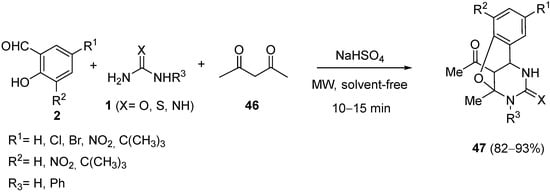
The synthesis of oxygen-bridged monastrol analogs
47 was reported by Cheng et al. using a Biginelli reaction of substituted salicylaldehydes
2, acetylacetone (
46), and urea or thiourea
1 with NaHSO
4 as the catalyst under microwave irradiation and solvent-free conditions in a short time and with good yields (
Scheme 20)
[77].
Scheme 20.
Synthesis of bridged DHPMs 47
.
Similarly, the synthesis of this kind of oxygen-bridged monastrol analogs by a Biginelli reaction was recently reported using a natural acidic medium of
Averrhoa bilimbi extract (ABE) as an eco-friendly and economically cheap, non-toxic acidic catalytic media
[78]. The advantages of this process are excellent yields of the obtained products, versatility in handling substrates, reuse of the catalyst, use of no hazardous organic solvents, and minimization of waste or side products.
Ramos and coworkers described the synthesis, characterization, and application of a new ion-tagged recyclable iron catalyst to the Biginelli reaction
[79]. The synthesis of dihydropyrimidine derivatives was performed by using MAI·Fe
2Cl
7 (5 mol%), different aromatic and aliphatic aldehydes, urea or thiourea, and 1,3-dicarbonyl compounds at 80 °C for 2 h. In particular, the use of acetylacetone or dimedone as 1,3-dicarbonyl compounds led to the corresponding products in good yields; these products are potent Eg5 inhibitors and have potent activity against MCF-7 and MDA-MB-231 cells
[80].
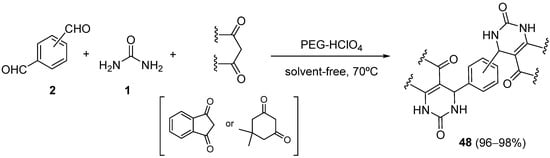
The use of dimedone or indane-1,3-dione as the 1,3 dicarbonyl compound in the Biginelli reaction was reported by Siddiqui et al. for the synthesis of novel bis-3,4-dihydropyrimidin-2(1
H)-one derivatives
48 in excellent yields using perchloric acid-modified poly(ethylene)glycol 6000 (PEG-HClO
4) as a biodegradable and reusable catalyst at ambient temperature under solvent-free condition at 70 °C (
Scheme 21)
[81].
Scheme 21.
Synthesis of bis-DHPMs 48
from dimedone or indane-1,3-dione as the keto ester component.
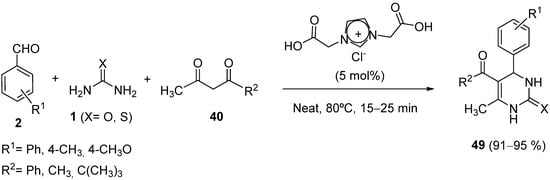
Recently, Davanagere and coworkers have reported a modified procedure for the synthesis of 1,3-bis(carboxymethyl)imidazolium chloride [BCMIM][Cl], a metal-free ionic catalyst, and its application in the one-pot multicomponent Biginelli reactions to dihydropyrimidine-2(1
H)-ones/thiones
49 in solvent-free conditions (
Scheme 22)
[82].
Scheme 22.
Synthesis of DHPMs 49
in ionic liquid.
A recent example of the use of acetylacetone as the 1,3-dicarbonyl compound in the Biginelli reaction is the synthesis of 5-acetyl-6-methyl-4-(1,3-diphenyl-1
H-pyrazol-4-yl)-3,4-dihydropyrmidin- 2(1
H)-thione, that was achieved in high yield by one-pot three-component synthesis using CaCl
2 in refluxing EtOH
[83]; this compound was used by the authors as starting material to synthesize a new series of 5-pyrazolyl; isoxazolyl; pyrimidinyl derivatives and also fused isoxazolo[5,4-
d]pyrimidine and pyrazolo[3,4-
d]pyrimidine; these compounds were evaluated for their antibacterial, antifungal and anti-inflammatory activity.
4.2. Keto Amides as the Keto Ester Component
4.2.1. Barbituric Acid Derivatives
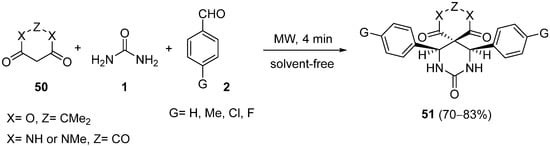
Shaabani and coworkers
[84][85][84,85] described the efficient synthesis of spiro-fused heterocycles using conventional heating
[85] or microwave irradiation
[84] under solvent-free conditions. The microwave-assisted one-pot method involves the heating of a mixture of Meldrum’s acid or barbituric acid derivatives
50 (instead of open chain cyclic β-dicarbonyl compounds), urea
1 and an aromatic aldehyde
2 in the presence of a protic acid catalyst to give a series of novel heterobicyclic compounds
51 in good yields and in a stereoselective manner (
Scheme 23). The best catalyst for the formation of the spiro-fused compounds was found to be acetic acid or NaHSO
4.
Scheme 23.
Synthesis of spiro-DHPMs 51
using Meldrum’s acid or barbituric acid derivatives 50
as the keto ester component.
Similarly, Mohammadi and coworkers reported the synthesis of spiropyrimidinethiones/spiropyrimidinones-barbituric acid derivatives
[86]. The one-pot reaction of barbituric acid, different benzaldehydes and urea or thiourea in the presence of a nanoporous acid catalyst of SBA-Pr-SO
3H, under solvent-free conditions, afforded novel heterobicyclic compounds in good yields. The spiro compounds were tested for their urease inhibitory activity against Jack bean urease
[87].
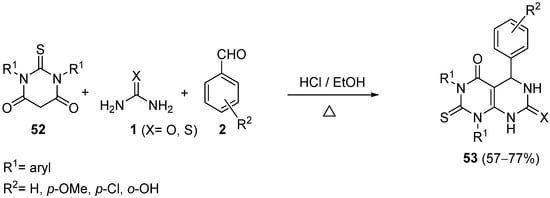
The use of thiobarbituric acid derivatives
52 was later reported by Dabholkar et al.
[87] for the Biginelli reaction with aromatic aldehydes
2 and urea or thiourea
1 using a catalytic amount of concentrated HCl in refluxing ethanol (
Scheme 24). Representative samples of the synthesized compounds
53 were screened for their anti-microbial activity.
Scheme 24.
Synthesis of DHMPs 53
using thiobarbituric acid derivatives 52
as the keto ester component.
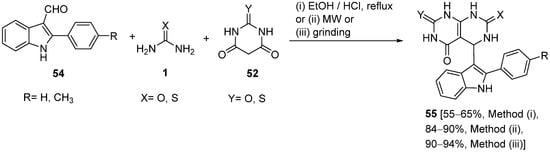
The synthesis of a series of 5-indolylpyrimido[4,5-
d]pyrimidinones
55 was reported by Gupta and coworkers
[88] by means of a multi-component reaction of 3-formylindoles
54, thiobarbituric acid/barbituric acid
52 and thiourea/urea (
1) in dry media (
Scheme 25). The reaction proceeded under conventional heating, microwave irradiation (MW), or grinding together neat reactants to give the titled compounds good-to-high yields. Representative compounds were also evaluated for their antimicrobial activity.
Scheme 25.
Synthesis of indolyl pyrimido pyrimidinones 55
using (thio)barbituric acid 52
as the keto ester component.
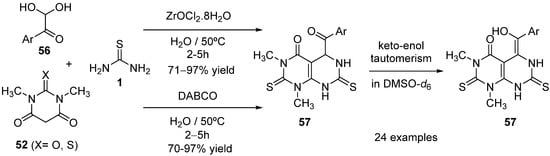
The synthesis of substituted pyrimido[4,5-d]pyrimidinones
57 using a Biginelli-like reaction was reported by Rimaz et al.
[89]; this transformation proceeds through a three-component tandem annulation of arylglyoxalmonohydrates
56 with 1,3-dimethylbarbituric acid
52 and thiourea
1 in the presence of catalytic amounts of 1,4-diazabicyclo[2.2.2]octane (DABCO) or L-proline. Later on, the same authors described the one-pot regioselective and chemoselective synthesis of the above-mentioned derivatives in water using two green catalytic systems (ZrOCl
2·8H
2O and DABCO); the desired products were obtained in good to excellent yields (
Scheme 26)
[90].
Scheme 26.
Synthesis of pyrimidopyrimidinones 57
using barbituric acid derivative 52
as the keto ester component.
4.2.2. Beta-Ketoamides and Beta-Ketosulfonamides
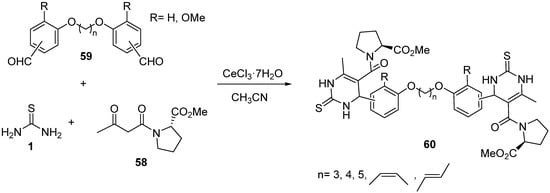
A series of conformationally flexible and restricted dimers of monastrol were described by Kamal and coworkers using a one-pot Biginelli multicomponent reaction
[91]. The β-keto amide intermediate
58, a derivative prepared from L-proline, was used as the 1,3-dicarbonyl compound in the Biginelli condensation with dibenzaldehydes
59 and thiourea
1 to obtain the asymmetric dimers
60 (
Scheme 27); these dimers were evaluated for cytotoxic potency against selected human cancer cell lines and some of the compounds exhibited more cytotoxic potency than the parent monastrol. In addition, the DNA binding ability and antimicrobial activities of these compounds were also evaluated, but with little success.
Scheme 27.
Synthesis of monastrol dimers 60
using proline-derived ketoester 58
as the keto ester component.
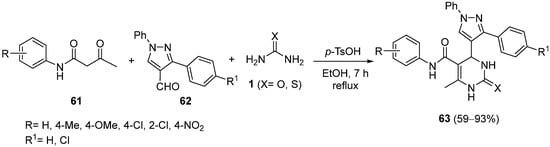
The synthesis of diarylpyrazole-ligated dihydropyrimidines possessing a lipophilic carbamoyl group
63 was reported by Yadlapalli et al.
[92]. The use of acetoacetanilide derivatives
61 as the 1,3-dicarbonyl compound in the Biginelli reaction with 1,3-diaryl-1
H-pyrazole-4-carbaldehydes
62 and urea/thiourea
1, afforded the corresponding dihydropyrimidine derivatives
63 with good-to-high yield (
Scheme 28); these novel compounds showed moderate anticancer activity against MCF-7 breast cancer cell lines as well as good to excellent antitubercular activity against MTB H37Rv.
Scheme 28.
Synthesis of pyrrolopyrimidinones 63
from ketoamides 61
as the keto ester component.
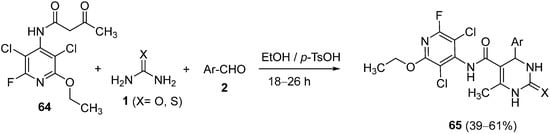
A series of novel 1,2,3,4-tetrahydropyrimidine derivatives were synthesized, in moderate to good yields, by Elumalai et al.
[93] by reacting
N-(3,5-dichloro-2-ethoxy-6-fluoropyridin-4-yl)-3-oxobutanamide
64, urea or thiourea
1 and aromatic aldehydes
2 in the presence of a catalytic amount of
p-toluen sulfonic acid (
p-TsOH) (
Scheme 29). The newly synthesized compounds
65 were evaluated for their antimycobacterial activity against
Mycobacterium tuberculosis.
Scheme 29.
Synthesis of DMPMs 65
using aryl acetamides 64
as the keto ester component.
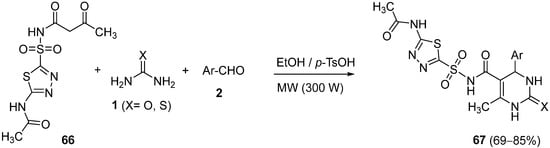
Later on, the same authors published the use of acetazolamide derived ketoamide
66 as a substrate for the Biginelli condensation with urea or thiourea
1 and aromatic aldehydes
2 under microwave irradiation (
Scheme 30)
[94]. The synthesized compounds
67 were evaluated for in vitro antimicrobial and cytotoxicity against
Bacillus subtilis,
Escherichia coli and
Vero cells.
Scheme 30.
Synthesis of DHPMs 67
using sulfonylacetamides 66
as the keto ester component.
More recently, these authors published the synthesis, antimicrobial activity, and in vitro cytotoxicity of novel sulphanilamide condensed 1,2,3,4-tetrahydropyrimidines
[95].
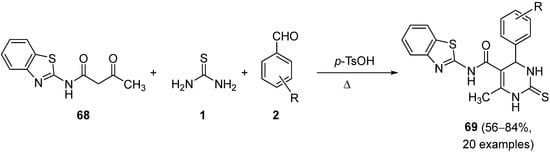
Chikhale et al. described the synthesis of novel derivatives of benzothiazolyl pyrimidine-5-carboxamides
69 which were synthesised by an acid-catalyzed one-pot three-component reaction of benzothiazolyl oxobutanamide
68, substituted aryl aldehydes
2 and thiourea
1 (
Scheme 31). The resulting products
69 were evaluated for their antitubercular activity to determine MIC against
Mycobacterium tuberculosis (H37Rv)
[96].
Scheme 31.
Synthesis of benzothiazolylpyrimidinones 69
using acetamides 68
as keto amide component.
Ramachandran and coworkers
[97] reported the syntheses of dihydropyrimidinones using the solvent-free grindstone chemistry method: a mixture of an aromatic aldehyde,
N-phenylacetoacetamide, urea/thiourea, cupric chloride, and a few drops of concentrated HCl was ground together to give the desired dihydropyrimidinones. The products were studied for their antibacterial activity.
Likewise, Gein and coworkers described the synthesis of 1,2,3,6-tetrahydro-pyrimidine-5-carboxamides by reacting arylacetoacetamides with aromatic aldehydes and urea under solvent-free conditions at 120–150 °C for 5–7 min
[98]. Good yields of the target compounds were obtained and the study of their antimicrobial activity was reported.
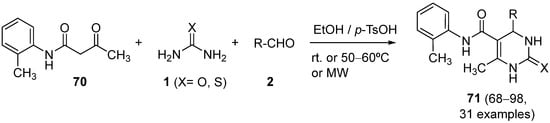
Recently, the parallel synthesis of new Biginelli 1,4-dihydropyrimidines
71 was reported by Faizan et al.
[99]. The desired compounds were synthesized via parallel synthesis by multicomponent-cyclisation reaction between aliphatic, aryl, heteroaryl aldehydes,
o-methyl acetoacetanilide
70, and excess of urea or thiourea
1 in absolute ethanol and using
p-toluen sulfonic acid as catalyst (
Scheme 32).
Scheme 32.
Synthesis of DHPMs 71
from acetamides 70
as the keto ester component.
Good yields were obtained and evaluation of anticancer activity and structure-activity relationships via 3D QSAR studies were carried out on the products.
4.3. Keto Acids as the Keto Ester Component
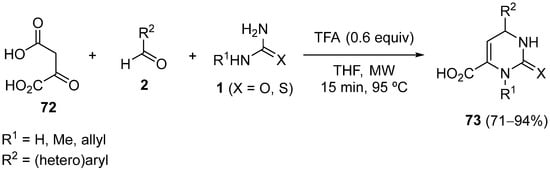
The use of β-keto carboxylic acids as substrates in the Biginelli reaction is scarce, given that a typical β-keto carboxylic acid should undergo spontaneous decarboxylation to give carbon dioxide and a ketone under the standard acidic reaction conditions; however, oxalacetic acid
72 does not undergo decomposition, presumably because the enol form is stabilized by resonance for both acids. Thus, replacing alkyl acetoacetate with oxalacetic acid
72 as a substrate for the Biginelli reaction led to the formation of 5-unsubstituted 3,4-dihydropyrimidin-2-(thio)-ones
73 due to in situ decarboxylation after cyclization
[100]. The major drawback of this reaction is the long reaction time (12 h); however, it can be conducted expeditiously with good yield and applied to a variety of reagents under microwave irradiation (
Scheme 33)
[101].
Scheme 33.
Synthesis of 5-unsubstituted 3,4-dihydropyrimidin-2-(thio)ones 73
.
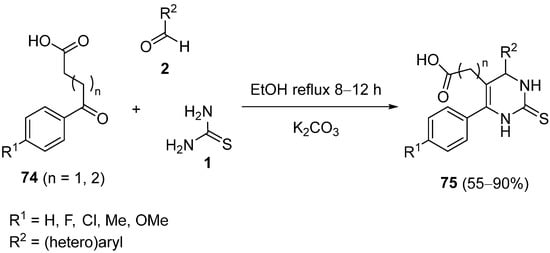
One special case of functionalized keto carboxylic acids is aromatic γ or δ-keto acids
74 (
Scheme 34); these are used as enolizable ketones in Biginelli-like reactions because aryl alkanoic acids are the most studied class of non-steroidal anti-inflammatory drugs (NSAIDs), such as diclofenac sodium, naproxen, ibuprofen, etc. Therefore, pyrimidine derivatives
75, with acetic or propanoic acid moiety at the fifth position, were synthesized to study their anti-inflammatory activity in vivo through the base-catalyzed condensation of aromatic γ or δ-keto acids
74, thiourea
1, and the appropriate aldehyde
2. Propanoic acid derivatives
75 (
n = 2) showed significant anti-inflammatory activity, due to their improved lipophilicity compare to acetic acid derivatives
75 (
n = 1)
[102][103][102,103].
Scheme 34.
Synthesis of pyrimidine derivatives 75
as non-steroidal anti-inflammatory drugs.
4.4. Ketones as the Keto Ester Component
In contrast to numerous protocols available for Biginelli reactions, Biginelli-like reactions using enolizable ketones instead of β-keto esters have been less explored
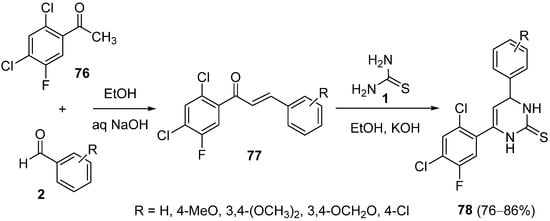
[104]. In 2004, Holla and coworkers synthesized 4,6-diaryl-3,4-dihydropyrimidin-2(1
H)-thiones
78 in a two-step protocol. Firstly, condensation of 2,4-dichloro-5-fluoroacetophenone
76 with benzaldehydes
2 under Claisen–Schmidt reaction conditions led to the corresponding chalcones
77. In a second step, these chalcones reacted with thiourea
1 in the presence of ethanolic potassium hydroxide to render final DHPMs
78 in good yields (
Scheme 35)
[105].
Scheme 35.
Two-step synthesis of 4,6-diaryl-3,4-dihydropyrimidin-2(1H
)-thiones 78.
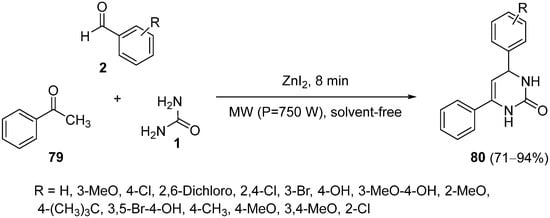
This type of reaction can be performed directly by the classical three-component one-pot synthesis with different systems. For instance, acetophenone
79 reacted with substituted benzaldehydes
2 and urea
1 in a microwave-assisted Biginelli-like reaction in a short and concise manner employing ZnI
2 as a catalyst under solvent-free conditions to afford DHPMs
80 (
Scheme 36)
[106]. The same reaction can be catalyzed by MnO
2/CNT(carbon nanotubes) nanocomposites with very good activity, recovery, and reusability of the catalyst
[107].
Scheme 36.
Microwave-assisted synthesis of 4,6-diaryl-3,4-dihydropyrimidin-2(1H
)-ones 80
.
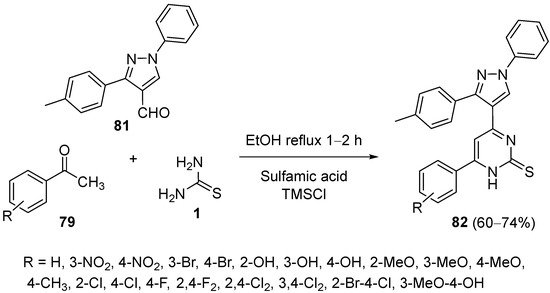
In 2020, Desai et al. reported a simple methodology for the synthesis of pyrimidinthione derivatives
82 via the condensation of substituted acetophenones
79, a pyrazol-4-carbaldehyde
81, thiourea
1 and sulfamic acid as the catalyst, in order to test their antimicrobial activity. TMSCl is believed to promote aromatization of the intermediates DHPMs (
Scheme 37)
[108].
Scheme 37.
Synthesis of pyrimidinthione derivatives 82
as antimicrobial agents.
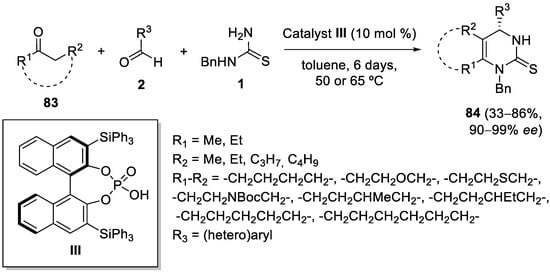
The first asymmetric catalytic version of Biginelli-like reactions using enolizable ketones as substrates was described by Li et al. in 2009
[104]; they found that BINOL-derived chiral organocatalyst
III was able to catalyze the reaction of cyclic and acyclic aliphatic ketones
83 with aromatic aldehydes
2 and
N-benzyl thiourea
1, yielding DHMPs
84 in excellent enantioselectivities (
Scheme 38). Aromatic ketones like 1-
p-tolylethanone gave only moderate enantioselectivity (61%
ee), and enolizable aliphatic aldehydes underwent a self-Biginelli-like reaction excluding the ketone from the reaction.
Scheme 38.
Enantioselective organocatalytic Biginelli-like condensations with cyclic and acyclic ketones.
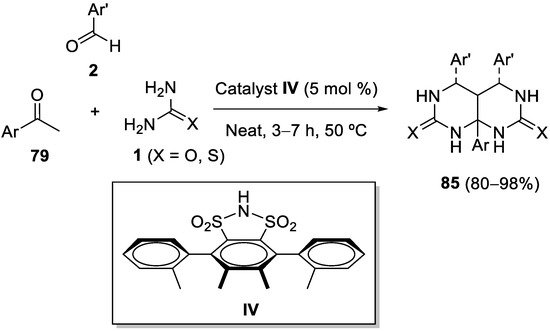
The chiral derivative of 1,2-benzenedisulfonimide
IV was found to be an efficient Brønsted acid catalyst to perform the standard Biginelli reaction of β-keto esters enantioselectively, with very high yields and excellent enantiomeric excesses. Surprisingly, when using acetophenones
79 as enolizable ketones instead of β-keto esters, two consecutive cyclizations occurred leading to the
meso form of adducts
85 in high yields (
Scheme 39); it seems that the nature of the acid catalyst and the absence of steric hindrances are decisive in leading the reaction towards this type of adducts
[109].
Scheme 39.
Diastereoselective synthesis of meso
adducts 85
.
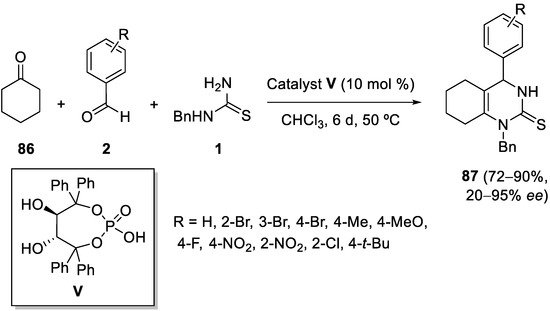
In the case of using cyclohexanone
86 as the enolizable ketone, the low cost and facile to prepare TADDOL-derived chiral phosphoric acid
V (obtained from natural tartaric acid) could be used to catalyze the Biginelli-like reaction with aromatic aldehydes
2 and
N-benzyl thiourea
1. The resulting enantioselectivity depends on the aldehyde substitution pattern (
Scheme 40)
[110].
Scheme 40.
Enantioselective organocatalytic Biginelli-like condensations with cyclohexanone.
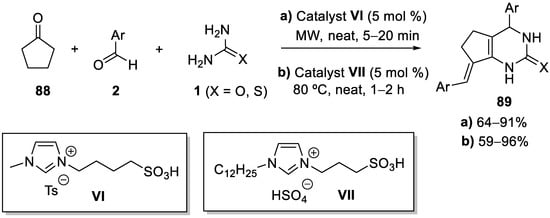
Unlike bigger cycloalkanones, cyclopentanone
88 has been described to furnish aryliden fused pyrimidinones
89 (
Scheme 41) through double α-reaction of the ketone, instead of the classical Biginelli-like product through single α-reaction
[111]; this transformation has been performed using different catalytic systems, most of them based on acidic ionic liquids (ILs). In the example shown in
Scheme 41 (conditions
a), Rahman et al. employed the Brønsted acid catalyst
VI under microwave irradiation and solvent-free conditions to produce heterobicyclic dihydropyrimidinone derivatives
89. The ionic liquid used as the catalyst could be reused at least six times without any noticeable decrease in catalytic activity. Attempts to expand the Biginelli-type reaction to condensations of cyclohexanone and/or aliphatic aldehydes lead to multiple unidentified products
[112]. Later on, the group of Professor Lu compared the efficiency of different Brønsted acidic ionic liquid catalysts in this transformation and concluded that the eco-friendly catalyst
VII gave the best results and could be reused at least seven times without significant loss of catalytic activity (
Scheme 41, conditions b)
[113].
Scheme 41.
Biginelli-like condensations of cyclopentanone catalyzed by acidic ionic liquids.
More examples of this transformation include the use of ILs immobilized in zeolites as catalysts
[114], or the use of AlCl
3 in poly(ethylene)glycol (PEG) as a green and reusable solvent
[115].
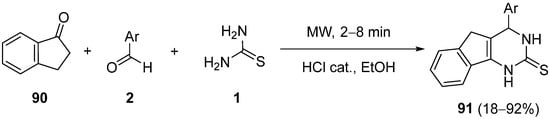
Aromatic cyclic ketones such as 1-indanone
90 (
Scheme 42) have also been used in Biginelli-type condensation with substituted benzaldehydes
2 and thiourea
1 to afford 4-aryl-1,3,4,5-tetrahydro-2
H-indeno[1,2-
d]pyrimidine-2-thiones
91 under microwave irradiation; these thiones were converted into their
S-alkylated/arylated derivatives in order to evaluate their antibacterial activities
[116].
Scheme 42.
Synthesis of indeno[1,2-d
]pyrimidine-2-thiones 91
under microwave irradiation.
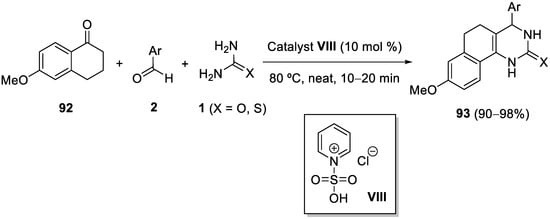
Similarly, fused DHPMs
93 were obtained by the condensation of 6-methoxy-1-tetralone
92, aromatic aldehydes
2, and urea or thiourea
1, in the presence of acidic IL
VIII as the catalyst under solvent-free conditions in excellent yields (
Scheme 43)
[117].
Scheme 43.
Biginelli-like condensations of 6-methoxy-1-tetralone catalyzed by acidic IL VIII
.
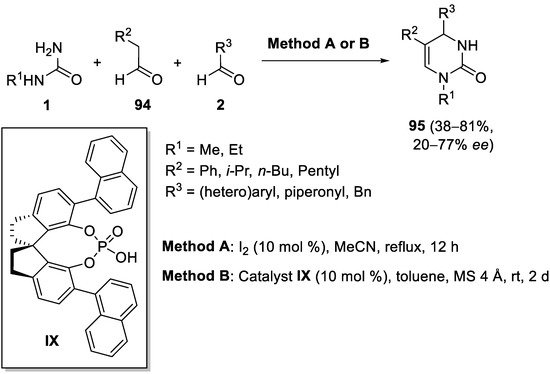
The utilization of enolizable aldehydes instead of ketones is scarce; however, in 2013 Qu et al. reported a highly chemo- and regio-selective tandem reaction of alkyl aldehydes
94, arylaldehydes
2 and mono-substituted urea
1, to give highly diverse 6-unsubstituted DHPMs
95 in reasonable yields under mild reaction conditions. The authors developed two different methods to carry out the reaction. Thus, method A (
Scheme 44) involved the use of molecular iodine as the catalyst, whereas method B represented the first catalytic enantioselective version of this reaction, by using chiral spirocyclic SPINOL-phosphoric acid
IX. Although the resulting enantioselectivities (
ee values) were from low-to-moderate (
Scheme 44)
[118].
Scheme 44.
Biginelli-type reaction of urea, alkyl aldehydes 94
, and aryl aldehydes.
A different family of 6-unsubstituted DHPMs
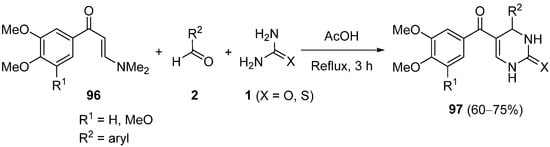
97 was prepared in 2016 by Bhat et al. using enaminones
96 as a surrogate of the enolizable carbonylic compound, with aromatic aldehydes
2 and urea or thiourea
1 in acetic acid (
Scheme 45); these DHPMs were evaluated for antitumor activity against cancer stem cells in vitro, and one of them (R
1 = MeO, R
2 = 4-EtOC
6H
4, X=O) demonstrated a remarkable antitumor effect in colon cancer xenografts in mice
[119].
Scheme 45.
Synthesis of 6-unsubstituted 3,4-dihydropyrimidin-2-(thio)ones 97
.
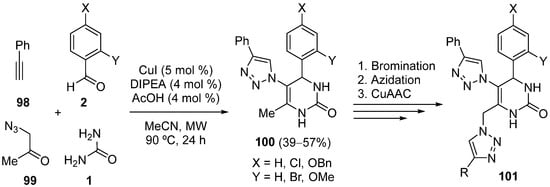
Another family of compounds with anticancer activity are the mono- and di(1,4-disubstituted 1,2,3-triazole)-DHPM hybrids
100 and
101, respectively (
Scheme 46). On the one hand, the monotriazole-DHPM hybrids
100 were synthesized by a one-pot multicomponent reaction involving a copper(I)-catalyzed alkyne-azide cycloaddition (CuAAC) and a Biginelly-like reaction, starting from phenylacetylene
98, 1-azidopropan-2-one
99, urea
1 and aromatic aldehydes
2. On the other hand, a multistep sequence of reactions that included bromination, azidation, and a CuAAC afforded the ditriazole-DHPM hybrids
101 [120].
Scheme 46.
Synthesis of mono- and di(1,4-disubstituted 1,2,3-triazole)-DHPM hybrids 100
and 101
.
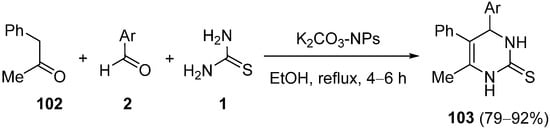
To conclude with the variants of ketones utilized as enolizable carbonylic compounds in Biginelli-like reactions, the condensation of phenylacetone
102, aromatic aldehydes
2, and thiourea
1 to give 4-aryl-5-phenyl-4-methyl substituted DHPMs
103, in the presence of potassium carbonate nanoparticles (NPs) to promote the reaction, was reported very recently; the antimicrobial activity of the synthesized compounds was also evaluated (
Scheme 47)
[121].
Scheme 47.
Synthesis of 4-aryl-5-phenyl-4-methyl substituted DHPMs 103.