Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Lamiaa M.A. Ali | -- | 2849 | 2022-08-02 10:13:44 | | | |
| 2 | Vivi Li | Meta information modification | 2849 | 2022-08-03 03:55:54 | | | | |
| 3 | Vivi Li | Meta information modification | 2849 | 2022-08-03 03:56:52 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Ali, L.M.A.; Gary-Bobo, M. Photochemical Internalization of siRNA for Cancer Therapy. Encyclopedia. Available online: https://encyclopedia.pub/entry/25762 (accessed on 23 July 2026).
Ali LMA, Gary-Bobo M. Photochemical Internalization of siRNA for Cancer Therapy. Encyclopedia. Available at: https://encyclopedia.pub/entry/25762. Accessed July 23, 2026.
Ali, Lamiaa Mohamed Ahmed, Magali Gary-Bobo. "Photochemical Internalization of siRNA for Cancer Therapy" Encyclopedia, https://encyclopedia.pub/entry/25762 (accessed July 23, 2026).
Ali, L.M.A., & Gary-Bobo, M. (2022, August 02). Photochemical Internalization of siRNA for Cancer Therapy. In Encyclopedia. https://encyclopedia.pub/entry/25762
Ali, Lamiaa Mohamed Ahmed and Magali Gary-Bobo. "Photochemical Internalization of siRNA for Cancer Therapy." Encyclopedia. Web. 02 August, 2022.
Copy Citation
In the race to design ever more effective therapy with ever more focused and controlled actions, nanomedicine and phototherapy seem to be two allies of choice. Indeed, the use of nanovectors making it possible to transport and protect genetic material is becoming increasingly important. In addition, the use of a method allowing the release of genetic material in a controlled way in space and time is also a strategy increasingly studied thanks to the use of lasers. In parallel, the use of interfering RNA and, more particularly, of small-interfering RNA (siRNA) has demonstrated significant potential for gene therapy.
nanovectors
photochemical internalization
siRNA
cancer
1. Introduction on Cancer and Treatments
Currently, cancer stands out as the first cause of death in the world after heart disease [1]. The increase in aging and population, as well as the changes in the distribution of the main risk factors, lead to rapid growth in cancer incidence and mortality. In 2020, 19.3 million new cases worldwide were identified, a number that is expected to increase to 28.4 million cases in 2040 [2].
Surgery, chemotherapy, radiotherapy, and hormone therapy are the main commonly used treatments despite the limitations of the specificity toward cancerous tissues, which lead to the key setbacks in cancer therapy as metastasis, tumor recurrence, and resistance to the treatments [3]. Therefore, there is an urgent need to develop new strategies to effectively kill cancer cells with little or no damage to healthy tissue.
Nanomedicine opens new hopes in solving many medical problems by developing several nanomaterials of organic or inorganic natures. The intrinsic properties of these nanomaterials, such as their nanometric size and large surface-to-volume ratio, open up many possibilities to explore their potential for the biomedical applications, especially for drug delivery, overcoming the chemotherapy limitations as systemic toxicity and multi-drug resistance mechanisms (MDR) [4].
Nowadays, several nanomedicines, a term that includes all nanomaterials used for biomedical applications [5], such as liposomes and albumin-based nanoparticles, are clinically approved for the treatment of cancer. Many others are in clinical trials and show great promises such as chemotherapy delivery systems, hyperthermia agents, and genetic or ribonucleic acid interference (RNAi) delivery systems [6].
2. Ribonucleic Acid Interference (RNAi) Technology
RNAi is a natural mechanism in eukaryotes for post-transcriptional gene silencing through (i) chromatin remodeling, (ii) inhibition of protein translation, or (iii) direct degradation of messenger RNA (mRNA) [7]. It was first discovered in 1998 by Fire and Mello research on Caenorhabditis elegans [8] and it serves as epigenetic regulator and defense mechanism against exogenous genes (e.g., viral or bacterial genes) and endogenous genes (e.g., transposons) [9][10][11]. In addition, it is considered as a promising strategy for treatment of cancer, primarily by specifically targeting key molecules involved in the molecular pathways of carcinogenesis [12][13]. RNAi mediates its action through non-coding short double-stranded RNA (nc-sdRNA) such as small-interfering RNA (siRNA) and microRNAs (miRNA). Single miRNA can inhibit the expression of several target genes simultaneously; however, to trigger gene silencing; siRNA is considered more efficient and specific than miRNA [14].
Here, researchers focus on siRNA; thus, a description of the mechanism of action, siRNA-based cancer therapies, and barriers to siRNA delivery will be discussed in the following paragraphs.
2.1. Mechanism of Action of siRNA
The biogenesis of siRNA starts with the presence of long dsRNA, which originates from different sources (e.g., viral, bacterial and synthetic RNA) in the cytoplasm (Figure 1). An enzyme called Dicer, a dsRNA-specific endoribonuclease from the RNase III protein family, cleaves the long dsRNA to about 21 nucleotides (nt) dsRNA called siRNA with 19 nt of complementary bases and a 2-nt overhang at each 3′-end. Afterwards, the formed siRNA duplex is loaded into a multiprotein RNA-induced silencing complex (RISC), in which a catalytic engine called the Argonaut protein (Ago-2) cleaves the passenger strand, keeping the active RISC with the guide strand. The siRNA guide strand recruits the RISC to complementary sequences in target mRNAs. A perfect siRNA base-pairing with mRNA causes direct mRNA cleavage by the catalytic RNase H domain of Ago-2, resulting in gene silencing, an effect that could last up to 7 days in rapidly divided cells and several weeks in nondividing cells [15][16].
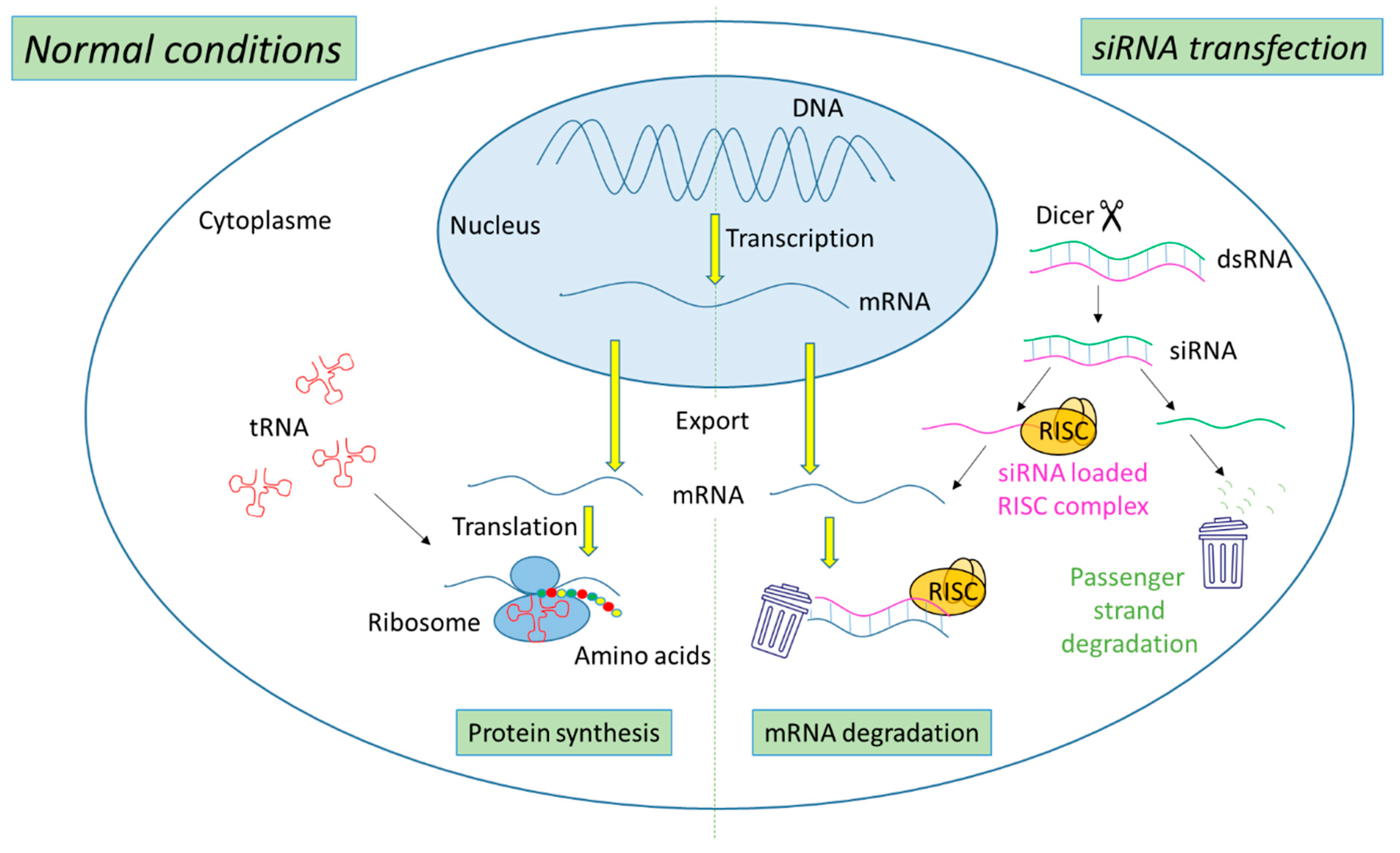
Figure 1. Representation of gene expression leading to protein synthesis in “normal conditions” in comparison with mechanism leading to mRNA degradation before protein synthesis in the presence of siRNA.
2.2. siRNA-Based Cancer Therapies
Recently, siRNA has emerged as a promising therapy for the treatment of several disorders, including cancer [17][18]. Its essential therapeutic strategy stems from its ability to suppress oncogenes and mutated tumor suppressor genes, as well as genes involved in MDR mechanism, resulting in the sensitization of cancer cells to treatment [19][20]. Anticancer siRNA targets can be categorized into (i) molecules involved in carcinogenesis, including molecules involved in oncogenic pathways, regulation of cell cycle, and apoptosis pathway; (ii) molecules involved in tumor–host interaction such as in cell adhesion, tumor extracellular matrix, tumor immune evasion, angiogenesis, invasion, and metastasis; and (iii) molecules participated in tumor resistance to chemotherapy, such as MDR and DNA repair proteins [14].
The first human clinical trial of siRNA encapsulated in targeted cyclodextrin polymer-based nanoparticles (CALAA-01) was started in 2008 by Calando Pharmaceuticals (Pasadena, CA, USA) for solid tumor cancer treatment. This phase I study was terminated in 2012 [21]. Table 1 summarizes siRNA-based cancer therapeutics in clinical trials.
Table 1. Anticancer siRNA-based therapeutics in clinical trials.
| Name/Sponsor | Route of Administration | Delivery System | Targeting Moiety | Target Gene | Disease | Clinical Trail Number (ClinicalTrials.gov) | Phase/Status | Period | Ref |
|---|---|---|---|---|---|---|---|---|---|
| CALAA-01/Calando Pharmaceuticals | i.v. | Cyclodextrin polymer-based nanoparticle | Transferrin | RRM2 | Solid tumors (Melanoma, gastrointestinal, prostate, etc.) | NCT00689065 | Phase I/Terminated | 2008–2012 | [21] |
| siG12D LODER/Silenseed Ltd. | Endoscopic intervention | Biodegradable Polymeric matrix | ----- | KRAS(G12D) and G12X mutations | Locally advanced pancreatic cancer | NCT01188785 | Phase I/Completed | 2011–2013 | [22] |
| siG12D-LODERs plus chemotherapy (Gemcitabine + nab-Paclitaxel or Folfirinox or modified Folfirinox) /Silenseed Ltd. | Endoscopic intervention | Biodegradable Polymeric matrix | ----- | KRAS(G12D) and G12X mutations | Locally advanced pancreatic cancer | NCT01676259 | Phase II/Recruiting | 2018–Est.2023 | [23] |
| ALN-VSP02/Alnylam Pharmaceuticals | i.v. | Lipid nanoparticle | ----- | VEGF KSP |
Solid tumors with liver involvement. | NCT00882180 NCT01158079 |
Phase I/Completed | 2009–2011 2010–2012 |
[24] |
| TKM-PLK1 (TKM-080301)/National Cancer Institute (NCI) | Hepatic Intra-Arterial Administration | Lipid nanoparticle | ----- | PLK1 | Primary or secondary liver cancer. | NCT01437007 | Phase I/Completed | 2011–2012 | [25] |
| Arbutus Biopharma Corporation | i.v. | Cancer, neuroendocrine tumors, adrenocortical carcinoma | NCT01262235 | Phase I/II/Completed | 2010–2015 | ||||
| Arbutus Biopharma Corporation | i.v. | Hepatocellular Carcinoma | NCT02191878 | Phase I/II/Completed | 2014–2016 | ||||
| DCR-MYC/Dicerna Pharmaceuticals, Inc. | i.v. | EnCoreTM lipid nanoparticle | ----- | MYC | Solid tumors, multiple myeloma, lymphoma | NCT02110563 | Phase I/Terminated | 2014–2016 | [26] |
| NBF-006/Nitto BioPharma, Inc. | Lipid nanoparticle | GSTP | Non-Small cell lung, pancreatic and colorectal Cancers | NCT03819387 | Phase I/Recruiting | 2019–Est.2023 | [27] | ||
| Atu027/Silence Therapeutics GmbH | i.v. | Liposomes | ----- | PKN3 | Advanced Solid Cancer | NCT00938574 | Phase I/Completed | 2009–2012 | [28] |
| Atu027-I-02 (Atu027 plus gemcitabine)/Silence Therapeutics GmbH | i.v. | Liposomes | ----- | PKN3 | Advanced or Metastatic Pancreatic Cancer | NCT01808638 | Phase I/II/Completed | 2013/2016 | [29] |
| EphA2-targeting DOPC-encapsulated siRNA/M.D. Anderson Cancer Center | i.v. | Liposomes | ----- | EphA2 | Advanced or recurrent solid tumors | NCT01591356 | Phase I/Active, not recruiting | 2015–Est.2024 | [30] |
| Mesenchymal Stromal Cells-derived Exosomes with KRAS(G12D) siRNA/M.D. Anderson Cancer Center | MSC exosome | CD47 | KRAS(G12D) | Metastatic pancreatic ductal adenocarcinoma with KrasG12D mutation | NCT03608631 | Phase I/Recruiting | 2021–Est.2023 | [31] |
RRM2: M2 subunit of ribonucleotide reductase; VEGF: vascular endothelial growth factor; KSP: kinesin spindle protein; PLK1: Polo-like kinase 1; PKN3: protein kinase N3; MYC: name of oncogene; DCR-MYC: anti-MYC DsiRNA formulated in EnCore lipid nanoparticles; EphA2: ephrin type-A receptor 2; DOPC: 1,2-dioleoyl-sn-glycero-3-phosphatidylcholine; KRAS(G12D): oncongene; MSC: mesenchymal stem cells; GSTP: glutathione-S-transferase P.
So far, only four non-cancer related siRNA-based therapeutics are approved by the Food and Drug administration (FDA), which are Patisiran, Givosiran, Lumasiran, and Inclisiran branded as ONPATTRO®, GIVLAARI®, OXLUMO®, and LEQVIO®, respectively, by Alnylam Pharmaceuticals (Cambridge, MD, USA) [32].
2.3. Hurdles to siRNA Delivery
The in vitro and in vivo delivery of “naked” siRNA, without a delivery system, can come up against several extracellular and intracellular obstacles such as the rapid degradation by nucleases (t½ ~ 10 min), rapid renal clearance, activation of the innate immune system, and the low accumulation in the target organ after systemic administration. Moreover, siRNA is characterized not only by low cellular uptake due to its negative charge and high molecular weight (~13 kDa) but also by its inability to escape from the endo-lysosomal compartments to the cytoplasm [33][34].
Thus, to circumvent these drawbacks two approaches are commonly used. The first approach is the chemical modification of the phosphate backbone, the heterocyclic nucleobase, or the ribose sugar moiety in order to increase siRNA stability, affinity, and specificity toward targets [35]. Three of the four FDA-approved siRNA therapeutics (Givosiran, Lumasiran and Inclisiran) are composed of chemically modified siRNA conjugated to trivalent N-acetylgalactosamine (GalNAc), a ligand to asialoglycoprotein receptor (ASGPR), resulting in hepatocyte-specific delivery. These GalNAc conjugates are fully modified at the 2′ position of the ribose sugar with 2′-O-methyl (2′-OMe) or 2′-deoxy-2′-fluoro (2′-F) as well as including phosphorothioate linkages. Unfortunately, chemical modifications are associated with several limitations, such as toxicity and low biological activity [36][37].
The second approach is the incorporation of siRNA into delivery systems to ensure efficient and safe administration of siRNA to the target site. For years, viral vectors have been used for siRNA delivery due to their strong efficiency, but they raise safety concerns due to their high immunogenicity and carcinogenic effects [38]. On the contrary, nanomaterials are considered as potential candidates for siRNA delivery showing low immunogenicity and toxicity, ease preparation, and high loading capacity. Additionally, the cargo is protected from degradation and nanomaterials can be active- or passive-targeted delivery systems, stimuli-responsive release systems, and co-delivery systems of different drugs simultaneously.
The first FDA-approved siRNA therapeutic, Patisiran, is composed of multicomponent lipid nanoparticles (LNP) encapsulating partially chemically modified siRNA, in which some of the nucleotides are chemically modified at 2′-OMe. These chemical modifications reduce the nuclease degradation and innate immune system stimulation, while LNP provides the liver-specific delivery of siRNA via apolipoprotein E (ApoE) receptor endocytosis aside from nuclease protection [39].
In general, nanomaterials are internalized in the cells by either nonendocytic or endocytic route depending on several factors such as nanomaterials physicochemical properties (e.g., size, shape, and charge); targeting moieties; etc. [40]. According to the mechanism of internalization, the fate of the nanomaterials inside cells is determined, for example if nanomaterials are internalized by clathrin-mediated endocytosis, then they will be trapped in the endosomes, which subsequently fuse with lysosomes and degradation will take place due to severe acidic conditions [4]. Therefore, the endo-lysosomal escape of nanomaterials for efficient cytosolic delivery of siRNA is mandatory in order to accomplish its biological activity.
Several strategies have been developed to enhance the cytosolic delivery of siRNA [41] such as proton sponge effect [42], fusogenic groups [43], and photochemical internalization (PCI) technology. This entry focuses on the PCI mechanism for siRNA release and the next paragraphs will present a description of this mechanism with several examples of PCI-mediated cytosolic delivery of siRNA using different vectors.
3. Photochemical Internalization (PCI) Mechanism
The PCI mechanism is a noninvasive technique that has developed over nearly two decades for multiple purposes including the treatment of cancer [44][45]. This technique is used to release macromolecules (peptides, proteins, and nucleic acids) confined in the endo-lysosomal compartments into the cytoplasm with the help of photosensitizers (PS) in light-dependent manner. Although, its similarity to photodynamic therapy (PDT) in components, including PS, oxygen, and light, differs from PDT in the final impact on cells. The PDT leads to cell death due to excessive production of reactive oxygen species (ROS), mainly singlet oxygen (1O2), which has a diffusion range of ~10–20 nm and t½ in µs [46][47][48]. While, PCI leads to disruption of endo-lysosomal membranes with no cytotoxic effect, as the accumulation of the PS in the endo-lysosomal membrane leads to local production of 1O2; hence, the damage is limited to its production zone [49].
The PCI process was first described by Berg K. et al. in 1999 [50] using several PS, including aluminum phtalocyanine disulfonate (AIPcS2a), in order to show their efficiency for the cytosolic delivery of plasmid encoding green fluorescent protein (GFP) into human colon cancer cells (HCT-116) and human melanoma cells (THX) after exposure to red light. In the study, they established the concept of PCI as an ideal site-specific delivery tool that could be combined with other therapeutic modalities [50]. Two years later, Berg team showed the potential of PCI mechanism for in vivo applications using AlPcS2a for the PCI delivery of gelonin in tumor-bearing mice [51]. In addition, AlPcS2a-based PCI delivery of bleomycin in tumors has also been reported [52]. In 2009, the first-in-man dose-escalating trial of PCI for bleomycin delivery in patients with different types of solid malignancies has started (phase 1, NCT00993512, ClinicalTrials.gov). The trial ended with the results demonstrating the safety of the photosensitizer used for PCI, which is Amphinex, a disulfonate tetraphenyl chlorin (TPCS2a) illuminated by 652-nm laser light with an energy of 60 J/cm2 [53].
Here, several siRNA vectors of different natures (lipid-based, polymer-based, peptide-based, and nanoparticles), which release their cargo under PCI mechanism, will be discussed (Figure 2).
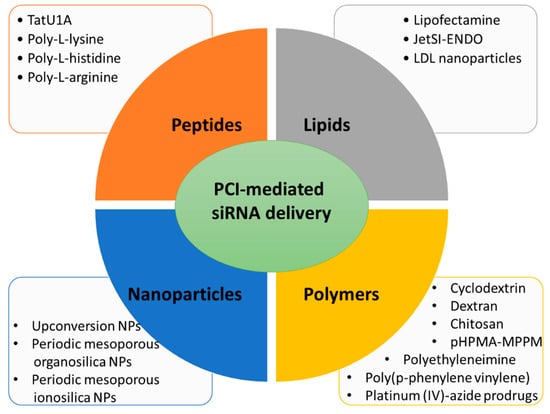
Figure 2. Main types of carriers used for PCI-mediated siRNA delivery discussed in this entry.
3.1. Lipid Carriers for PCI-Mediated siRNA Delivery
The first evidence that PCI induces endo-lysosomal escape of siRNA was a paper published in 2007 by Oliveira S. and co-workers [54]. In this work the proof of concept was performed by using siRNA directed against epidermal growth factor receptor (EGFR), a molecular target of several cancers, complexed with lipofectamineTM. Human epidermoid carcinoma cells (A431) in culture were incubated with this complex (lipofectamine/siRNA) and a photosensitizer, meso-tetraphenylporphyrin disulfonate (TPPS2a), necessary to destabilize the endo-lysosomal membranes under photoactivation, leading to the PCI mechanism. Light excitation (wavelength at 375–450 nm) demonstrated the efficiency for lysosomal escape of the complex lipofectamine/anti-EGFR siRNA by increasing the knockdown of the EGFR protein expression level. However, the cytotoxicity generated by lipofectamine and its efficiency even without photoexcitation limit any in vivo use [55]. This characteristic has led Boe S. and coworkers to perform PCI of siRNA, using safer lipid carriers [56]. In their work, authors chose the cationic lipid jetSI-ENDO to complex siRNA against S100A4, a protein responsible for invasive and metastatic phenotype in cancer. The TPPS2a has been used as photosensitizer to destabilize the endosomal membranes and allow PCI. In this work, the high silencing efficiency was demonstrated by a dramatic decrease in mRNA and protein expression levels after light excitation. Even if this system is very powerful, it remains relatively complex because, here too, the authors must manipulate several components. Indeed, they must add a PS to their cationic support to deliver siRNA, which can be delivered under light excitation. It is also the case in the work demonstrating the possible use of low density lipoprotein (LDL) nanoparticle for siRNA delivery [57]. Here, siRNA was conjugated to cholesterol and could then be encapsulated in LDL nanoparticles. The efficiency of mRNA knockdown was around 38% and reached 78% when applying PCI with AlPcS2a at 660 nm.
In the three discussed examples, cells were preincubated with the PS followed by the addition of the lipoplexes, although they showed a high transfection capacity (70–90%), an all-in-one carrier is necessary for ease of handing. Additionally, in term of toxicity, the model of LDL nanoparticles is safer than the nonmetabolized lipofectamineTM or JetSITM and could be introduced in in vivo system. Finally, using red light irradiation is favorable in terms of phototoxicity and penetration depth.
3.2. Peptide Carriers for PCI-Mediated siRNA Delivery
The cell-penetrating peptides (CPPs) are high potent tools to enable (macro)molecules delivery in mammalian cells [58][59]. Endoh T. et al. elaborated a molecular construction consisting of a complexation of TatU1A (fusion of TAT peptide with U1A RNA binding domain) with a fluorophore (Alexa Fluor 546) and a siRNA associated to U1A RNA binding domain (U1AsiRNA) [60]. This macromolecule was well-internalized via the endo-lysosomal pathway of the mammalian cells used in the study, the Chinese Hamster Ovary (CHO) cells. Among the various strategies known to destabilize the endo-lysosomal membranes for a lysosomal escape, mainly drugs, the photostimulation of the fluorophores was already described as an efficient, precise and controlled mechanism [61][62]. Here, the high efficiency of the cytosolic delivery of the siRNA carried by a CPP complex was demonstrated by the photo-stimulation with Alexa Fluor 546 (60 s, 540 nm, 100 Watt halogen lamp) allowing PCI and obtaining an effect of GFP gene silencing indicated by approximately 70% decrease in relative fluorescence intensity [60].
In the race for biosafety, biocompatibility and biodegradability of drug delivery systems and gene transporters, the polyamino acids family has demonstrated very interesting properties as well as high efficiency, whether modified to acquire or not proton sponge capacity for lysosomal escape [63]. Jorgensen J.A.L et al. showed for the first time the capacity of the unmodified poly-L-arginine, poly-L-histidine or poly-L-lysine to carry and deliver siRNA under PCI mechanism activated by blue light in the presence of TPPS2a as photosensitizer [63].
A number of CPPs-photosensitizers conjugates has been designed and used for PCI [64]. Conjugation of CPPs to TPP provides high quantum yield compared to that conjugated to Alexa546 or Alexa633 [65][66]. Unfortunately, translating this strategy from bench to bedside is limited due to the low bioavailability of CPPs and restricted biodistribution. In addition, the cell internalization of CPPs lacks the specificity and is sometimes restricted [67][68]. Peptides of arginine are precious tool for siRNA delivery by PCI as they lack the proton sponge property. In addition, they are internalized into cells more easily than peptides of lysine or histidine [69].
References
- Yahya, E.B.; Alqadhi, A.M. Recent trends in cancer therapy: A review on the current state of gene delivery. Life Sci. 2021, 269, 119087.
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA A Cancer J. Clin. 2021, 71, 209–249.
- Kesse, S.; Boakye-Yiadom, K.O.; Ochete, B.O.; Opoku-Damoah, Y.; Akhtar, F.; Filli, M.S.; Farooq, M.A.; Aquib, M.; Mily, B.J.M.; Murtaza, G. Mesoporous Silica Nanomaterials: Versatile Nanocarriers for Cancer Theranostics and Drug and Gene Delivery. Pharmaceutics 2019, 11, 77.
- Ali, L.M.A. Toxicity Studies of Polymer Based Supermagnetic Iron Oxide Nanoparticles. Ph.D. Thesis, Universidad Zaragoza, Zaragoza, Spain, 2014.
- Duncan, R.; Gaspar, R. Nanomedicine(s) under the microscope. Mol. Pharm. 2011, 8, 2101–2141.
- Shi, J.; Kantoff, P.W.; Wooster, R.; Farokhzad, O.C. Cancer nanomedicine: Progress, challenges and opportunities. Nat. Rev. Cancer 2017, 17, 20–37.
- Mansoori, B.; Shotorbani, S.S.; Baradaran, B. RNA Interference and its Role in Cancer Therapy. Adv. Pharm. Bull. 2014, 4, 313–321.
- Fire, A.; Xu, S.; Montgomery, M.K.; Kostas, S.A.; Driver, S.E.; Mello, C.C. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 1998, 391, 806–811.
- Tan, F.L.; Yin, J.Q. RNAi, a new therapeutic strategy against viral infection. Cell Res. 2004, 14, 460–466.
- Holoch, D.; Moazed, D. RNA-mediated epigenetic regulation of gene expression. Nat. Rev. Genet. 2015, 16, 71–84.
- Hammond, S.M.; Caudy, A.A.; Hannon, G.J. Post-transcriptional gene silencing by double-stranded RNA. Nat. Rev. Genet. 2001, 2, 110–119.
- Yoon, J.; Shin, M.; Lee, J.Y.; Lee, S.N.; Choi, J.H.; Choi, J.W. RNA interference (RNAi)-based plasmonic nanomaterials for cancer diagnosis and therapy. J. Control. Release 2022, 342, 228–240.
- Pai, S.; Lin, Y.Y.; Macaes, B.; Meneshian, A.; Hung, C.F.; Wu, T.C. Prospects of RNA interference therapy for cancer. Gene Ther. 2006, 13, 464–477.
- Pengnam, S.; Plianwong, S.; Yingyongnarongkul, B.-k.; Patrojanasophon, P.; Opanasopit, P. Delivery of small interfering RNAs by nanovesicles for cancer therapy. Drug Metab. Pharmacokinet. 2022, 42, 100425.
- Miele, E.; Spinelli, G.P.; Miele, E.; Di Fabrizio, E.; Ferretti, E.; Tomao, S.; Gulino, A. Nanoparticle-based delivery of small interfering RNA: Challenges for cancer therapy. Int. J. Nanomed. 2012, 7, 3637–3657.
- Kim, D.H.; Rossi, J.J. Strategies for silencing human disease using RNA interference. Nat. Rev. Genet. 2007, 8, 173–184.
- Lares, M.R.; Rossi, J.J.; Ouellet, D.L. RNAi and small interfering RNAs in human disease therapeutic applications. Trends Biotechnol. 2010, 28, 570–579.
- de Fougerolles, A.; Vornlocher, H.P.; Maraganore, J.; Lieberman, J. Interfering with disease: A progress report on siRNA-based therapeutics. Nat. Rev. Drug Discov. 2007, 6, 443–453.
- Dönmez, Y.; Gündüz, U. Reversal of multidrug resistance by small interfering RNA (siRNA) in doxorubicin-resistant MCF-7 breast cancer cells. Biomed. Pharmacother. 2011, 65, 85–89.
- Singh, A.; Trivedi, P.; Jain, N.K. Advances in siRNA delivery in cancer therapy. Artif. Cells Nanomed. Biotechnol. 2017, 46, 274–283.
- Zuckerman, J.E.; Gritli, I.; Tolcher, A.; Heidel, J.D.; Lim, D.; Morgan, R.; Chmielowski, B.; Ribas, A.; Davis, M.E.; Yen, Y. Correlating animal and human phase Ia/Ib clinical data with CALAA-01, a targeted, polymer-based nanoparticle containing siRNA. Proc. Natl. Acad. Sci. USA 2014, 111, 11449–11454.
- Golan, T.; Khvalevsky, E.Z.; Hubert, A.; Gabai, R.M.; Hen, N.; Segal, A.; Domb, A.; Harari, G.; Ben David, E.; Raskin, S.; et al. RNAi therapy targeting KRAS in combination with chemotherapy for locally advanced pancreatic cancer patients. Oncotarget 2015, 6, 24560–24570.
- Varghese, A.M.; Ang, C.; DiMaio, C.J.; Javle, M.M.; Gutierrez, M.; Yarom, N.; Stemmer, S.M.; Golan, T.; Geva, R.; Semenisty, V.; et al. A phase II study of siG12D-LODER in combination with chemotherapy in patients with locally advanced pancreatic cancer (PROTACT). J. Clin. Oncol. 2020, 38, TPS4672.
- Tabernero, J.; Shapiro, G.I.; Lorusso, P.M.; Cervantes, A.; Schwartz, G.K.; Weiss, G.J.; Paz-Ares, L.; Cho, D.C.; Infante, J.R.; Alsina, M.; et al. First-in-Humans Trial of an RNA Interference Therapeutic Targeting VEGF and KSP in Cancer Patients with Liver Involvement. Cancer Discov. 2013, 3, 406–417.
- Barba, A.A.; Bochicchio, S.; Dalmoro, A.; Lamberti, G. Lipid Delivery Systems for Nucleic-Acid-Based-Drugs: From Production to Clinical Applications. Pharmaceutics 2019, 11, 360.
- Tolcher, A.W.; Papadopoulos, K.P.; Patnaik, A.; Rasco, D.W.; Martinez, D.; Wood, D.L.; Fielman, B.; Sharma, M.; Janisch, L.A.; Brown, B.D.; et al. Safety and activity of DCR-MYC, a first-in-class Dicer-substrate small interfering RNA (DsiRNA) targeting MYC, in a phase I study in patients with advanced solid tumors. J. Clin. Oncol. 2015, 33, 11006.
- A Study of NBF-006 in Non-Small Cell Lung, Pancreatic, or Colorectal Cancer. Available online: https://clinicaltrials.gov/ct2/show/NCT03819387 (accessed on 3 May 2022).
- Schultheis, B.; Strumberg, D.; Santel, A.; Vank, C.; Gebhardt, F.; Keil, O.; Lange, C.; Giese, K.; Kaufmann, J.; Khan, M.; et al. First-in-Human Phase I Study of the Liposomal RNA Interference Therapeutic Atu027 in Patients With Advanced Solid Tumors. J. Clin. Oncol. 2014, 32, 4141–4148.
- Schultheis, B.; Strumberg, D.; Kuhlmann, J.; Wolf, M.; Link, K.; Seufferlein, T.; Kaufmann, J.; Feist, M.; Gebhardt, F.; Khan, M.; et al. Safety, Efficacy and Pharcacokinetics of Targeted Therapy with The Liposomal RNA Interference Therapeutic Atu027 Combined with Gemcitabine in Patients with Pancreatic Adenocarcinoma. A Randomized Phase Ib/IIa Study. Cancers 2020, 12, 3130.
- Oner, E.; Kotmakci, M.; Baird, A.M.; Gray, S.G.; Butuner, B.D.; Bozkurt, E.; Kantarci, A.G.; Finn, S.P. Development of EphA2 siRNA-loaded lipid nanoparticles and combination with a small-molecule histone demethylase inhibitor in prostate cancer cells and tumor spheroids. J. Nanobiotechnol. 2021, 19, 71.
- Lee, B.-C.; Kang, I.; Yu, K.-R. Therapeutic Features and Updated Clinical Trials of Mesenchymal Stem Cell (MSC)-Derived Exosomes. J. Clin. Med. 2021, 10, 711.
- Zhang, M.M.; Bahal, R.; Rasmussen, T.P.; Manautou, J.E.; Zhong, X.-B. The growth of siRNA-based therapeutics: Updated clinical studies. Biochem. Pharmacol. 2021, 189, 114432.
- Liu, F.; Shollenberger, L.; Conwell, C.C.; Yuan, X.; Huang, L. Mechanism of naked DNA clearance after intravenous injection. J. Gene Med. 2007, 9, 613–619.
- Kanasty, R.L.; A Whitehead, K.; Vegas, A.J.; Anderson, D.G. Action and Reaction: The Biological Response to siRNA and Its Delivery Vehicles. Mol. Ther. 2012, 20, 513–524.
- Sharma, V.K.; Sharma, R.K.; Singh, S.K. Antisense oligonucleotides: Modifications and clinical trials. MedChemComm 2014, 5, 1454–1471.
- Nair, J.K.; Willoughby, J.L.S.; Chan, A.; Charisse, K.; Alam, R.; Wang, Q.; Hoekstra, M.; Kandasamy, P.; Kel’In, A.V.; Milstein, S.; et al. Multivalent N-Acetylgalactosamine-Conjugated siRNA Localizes in Hepatocytes and Elicits Robust RNAi-Mediated Gene Silencing. J. Am. Chem. Soc. 2014, 136, 16958–16961.
- Gangopadhyay, S.; Gore, K.R. Advances in siRNA therapeutics and synergistic effect on siRNA activity using emerging dual ribose modifications. RNA Biol. 2022, 19, 452–467.
- Thomas, C.E.; Ehrhardt, A.; Kay, M.A. Progress and problems with the use of viral vectors for gene therapy. Nat. Rev. Genet. 2003, 4, 346–358.
- Mikami, A.; Erande, N.; Matsuda, S.; Kel’in, A.; Woods, L.B.; Chickering, T.; Pallan, P.S.; Schlegel, M.K.; Zlatev, I.; Egli, M.; et al. Synthesis, chirality-dependent conformational and biological properties of siRNAs containing 5′-(R)- and 5′-(S)-C-methyl-guanosine. Nucleic Acids Res. 2020, 48, 10101–10124.
- Nel, A.E.; Mädler, L.; Velegol, D.; Xia, T.; Hoek, E.M.V.; Somasundaran, P.; Klaessig, F.; Castranova, V.; Thompson, M. Understanding biophysicochemical interactions at the nano–bio interface. Nat. Mater. 2009, 8, 543–557.
- Guo, S.; Huang, L. Nanoparticles Escaping RES and Endosome: Challenges for siRNA Delivery for Cancer Therapy. J. Nanomater. 2011, 2011, 1–12.
- Yezhelyev, M.V.; Qi, L.; O’Regan, R.M.; Nie, S.; Gao, X. Proton-Sponge Coated Quantum Dots for siRNA Delivery and Intracellular Imaging. J. Am. Chem. Soc. 2008, 130, 9006–9012.
- Hoffmann, M.; Hersch, N.; Merkel, R.; Csiszar, A.; Hoffmann, B. Changing the Way of Entrance: Highly Efficient Transfer of mRNA and siRNA via Fusogenic Nano-Carriers. J. Biomed. Nanotechnol. 2019, 15, 170–183.
- Otterhaug, T.; Janetzki, S.; Welters, M.J.P.; Håkerud, M.; Nedberg, A.G.; Edwards, V.T.; Boekestijn, S.; Loof, N.M.; Selbo, P.K.; Olivecrona, H.; et al. Photochemical Internalization Enhanced Vaccination Is Safe, and Gives Promising Cellular Immune Responses to an HPV Peptide-Based Vaccine in a Phase I Clinical Study in Healthy Volunteers. Front. Immunol. 2021, 11, 576756.
- Dechêne, A.; Kasper, S.; Olivecrona, H.; Schirra, J.; Trojan, J. Photochemical internalization and gemcitabine combined with first-line chemotherapy in perihilar cholangiocarcinoma: Observations in three patients. Endosc. Int. Open 2020, 8, E1878–E1883.
- Ożog, L.; Aebisher, D. Singlet oxygen lifetime and diffusion measurements. Eur. J. Clin. Exp. Med. 2018, 16, 123–126.
- Klaper, M.; Fudickar, W.; Linker, T. Role of Distance in Singlet Oxygen Applications: A Model System. J. Am. Chem. Soc. 2016, 138, 7024–7029.
- Raemdonck, K.; Naeye, B.; Høgset, A.; Demeester, J.; De Smedt, S.C. Prolonged gene silencing by combining siRNA nanogels and photochemical internalization. J. Control. Release 2010, 145, 281–288.
- Berg, K.; Folini, M.; Prasmickaite, L.; Selbo, P.; Bonsted, A.; Engesaeter, B.; Zaffaroni, N.; Weyergang, A.; Dietzea, A.; Maelandsmo, G.; et al. Photochemical Internalization: A New Tool for Drug Delivery. Curr. Pharm. Biotechnol. 2007, 8, 362–372.
- Berg, K.; Selbo, P.K.; Prasmickaite, L.; E Tjelle, T.; Sandvig, K.; Moan, J.; Gaudernack, G.; Fodstad, O.; Kjølsrud, S.; Anholt, H.; et al. Photochemical internalization: A novel technology for delivery of macromolecules into cytosol. Cancer Res. 1999, 59, 1180–1183.
- Selbo, P.K.; Sivam, G.; Fodstad, O.; Sandvig, K.; Berg, K. In vivo documentation of photochemical internalization, a novel approach to site specific cancer therapy. Int. J. Cancer 2001, 92, 761–766.
- Berg, K.; Dietze, A.; Kaalhus, O.; Høgset, A. Site-Specific Drug Delivery by Photochemical Internalization Enhances the Antitumor Effect of Bleomycin. Clin. Cancer Res. 2005, 11, 8476–8485.
- Sultan, A.; Jerjes, W.; Berg, K.; Høgset, A.; Mosse, C.A.; Hamoudi, R.; Hamdoon, Z.; Simeon, C.; Carnell, D.; Forster, M.; et al. Disulfonated tetraphenyl chlorin (TPCS2a)-induced photochemical internalisation of bleomycin in patients with solid malignancies: A phase 1, dose-escalation, first-in-man trial. Lancet Oncol. 2016, 17, 1217–1229.
- Oliveira, S.; Fretz, M.M.; Høgset, A.; Storm, G.; Schiffelers, R.M. Photochemical internalization enhances silencing of epidermal growth factor receptor through improved endosomal escape of siRNA. Biochim. et Biophys. Acta (BBA)-Biomembr. 2007, 1768, 1211–1217.
- Kraja, I.; Bing, R.; Hiwatashi, N.; Rousseau, B.; Nalband, D.; Kirshenbaum, K.; Branski, R.C. Preliminary study of a novel transfection modality for in vivo siRNA delivery to vocal fold fibroblasts. Laryngoscope 2017, 127, E231–E237.
- Bøe, S.; Longva, A.; Hovig, E. Photochemically Induced Gene Silencing Using Small Interfering RNA Molecules in Combination with Lipid Carriers. Oligonucleotides 2007, 17, 166–173.
- Jin, H.; Lovell, J.F.; Chen, J.; Lin, Q.; Ding, L.; Ng, K.K.; Pandey, R.K.; Manoharan, M.; Zhang, Z.; Zheng, G. Mechanistic Insights into LDL Nanoparticle-Mediated siRNA Delivery. Bioconjug. Chem. 2011, 23, 33–41.
- Schwarze, S.R.; Ho, A.; Vocero-Akbani, A.; Dowdy, S.F. In Vivo Protein Transduction: Delivery of a Biologically Active Protein into the Mouse. Science 1999, 285, 1569–1572.
- Del Gaizo, V.; Payne, R.M. A novel TAT-mitochondrial signal sequence fusion protein is processed, stays in mitochondria, and crosses the placenta. Mol. Ther. 2003, 7, 720–730.
- Endoh, T.; Sisido, M.; Ohtsuki, T. Cellular siRNA Delivery Mediated by a Cell-Permeant RNA-Binding Protein and Photoinduced RNA Interference. Bioconjug. Chem. 2008, 19, 1017–1024.
- Maiolo, J.R.; Ottinger, E.A.; Ferrer, M. Specific Redistribution of Cell-Penetrating Peptides from Endosomes to the Cytoplasm and Nucleus upon Laser Illumination. J. Am. Chem. Soc. 2004, 126, 15376–15377.
- Matsushita, M.; Noguchi, H.; Lu, Y.-F.; Tomizawa, K.; Michiue, H.; Li, S.-T.; Hirose, K.; Bonner-Weir, S.; Matsui, H. Photo-acceleration of protein release from endosome in the protein transduction system. FEBS Lett. 2004, 572, 221–226.
- Jørgensen, J.A.L.; Longva, A.S.; Hovig, E.; Bøe, S.L. Evaluation of Biodegradable Peptide Carriers for Light-Directed Targeting. Nucleic Acid Ther. 2013, 23, 131–139.
- Soe, T.H.; Watanabe, K.; Ohtsuki, T. Photoinduced Endosomal Escape Mechanism: A View from Photochemical Internalization Mediated by CPP-Photosensitizer Conjugates. Molecules 2020, 26, 36.
- Wang, J.T.-W.; Giuntini, F.; Eggleston, I.M.; Bown, S.G.; MacRobert, A.J. Photochemical internalisation of a macromolecular protein toxin using a cell penetrating peptide-photosensitiser conjugate. J. Control. Release 2012, 157, 305–313.
- Ohtsuki, T.; Miki, S.; Kobayashi, S.; Haraguchi, T.; Nakata, E.; Hirakawa, K.; Sumita, K.; Watanabe, K.; Okazaki, S. The molecular mechanism of photochemical internalization of cell penetrating peptide-cargo-photosensitizer conjugates. Sci. Rep. 2015, 5, 18577.
- Sarko, D.; Beijer, B.; Garcia Boy, R.; Nothelfer, E.-M.; Leotta, K.; Eisenhut, M.; Altmann, A.; Haberkorn, U.; Mier, W. The Pharmacokinetics of Cell-Penetrating Peptides. Mol. Pharm. 2010, 7, 2224–2231.
- Krämer, S.; Wunderli-Allenspach, H. No entry for TAT(44–57) into liposomes and intact MDCK cells: Novel approach to study membrane permeation of cell-penetrating peptides. Biochim. et Biophys. Acta (BBA)-Biomembr. 2002, 1609, 161–169.
- Mitchell, D.; Steinman, L.; Kim, D.; Fathman, C.; Rothbard, J. Polyarginine enters cells more efficiently than other polycationic homopolymers. J. Pept. Res. 2000, 56, 318–325.
More
Information
Subjects:
Biology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.4K
Revisions:
3 times
(View History)
Update Date:
03 Aug 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No