In the race to design ever more effective therapy with ever more focused and controlled actions, nanomedicine and phototherapy seem to be two allies of choice. Indeed, the use of nanovectors making it possible to transport and protect genetic material is becoming increasingly important. In addition, the use of a method allowing the release of genetic material in a controlled way in space and time is also a strategy increasingly studied thanks to the use of lasers. In parallel, the use of interfering RNA and, more particularly, of small-interfering RNA (siRNA) has demonstrated significant potential for gene therapy.
- nanovectors
- photochemical internalization
- siRNA
- cancer
1. Introduction on Cancer and Treatments
2. Ribonucleic Acid Interference (RNAi) Technology
2.1. Mechanism of Action of siRNA
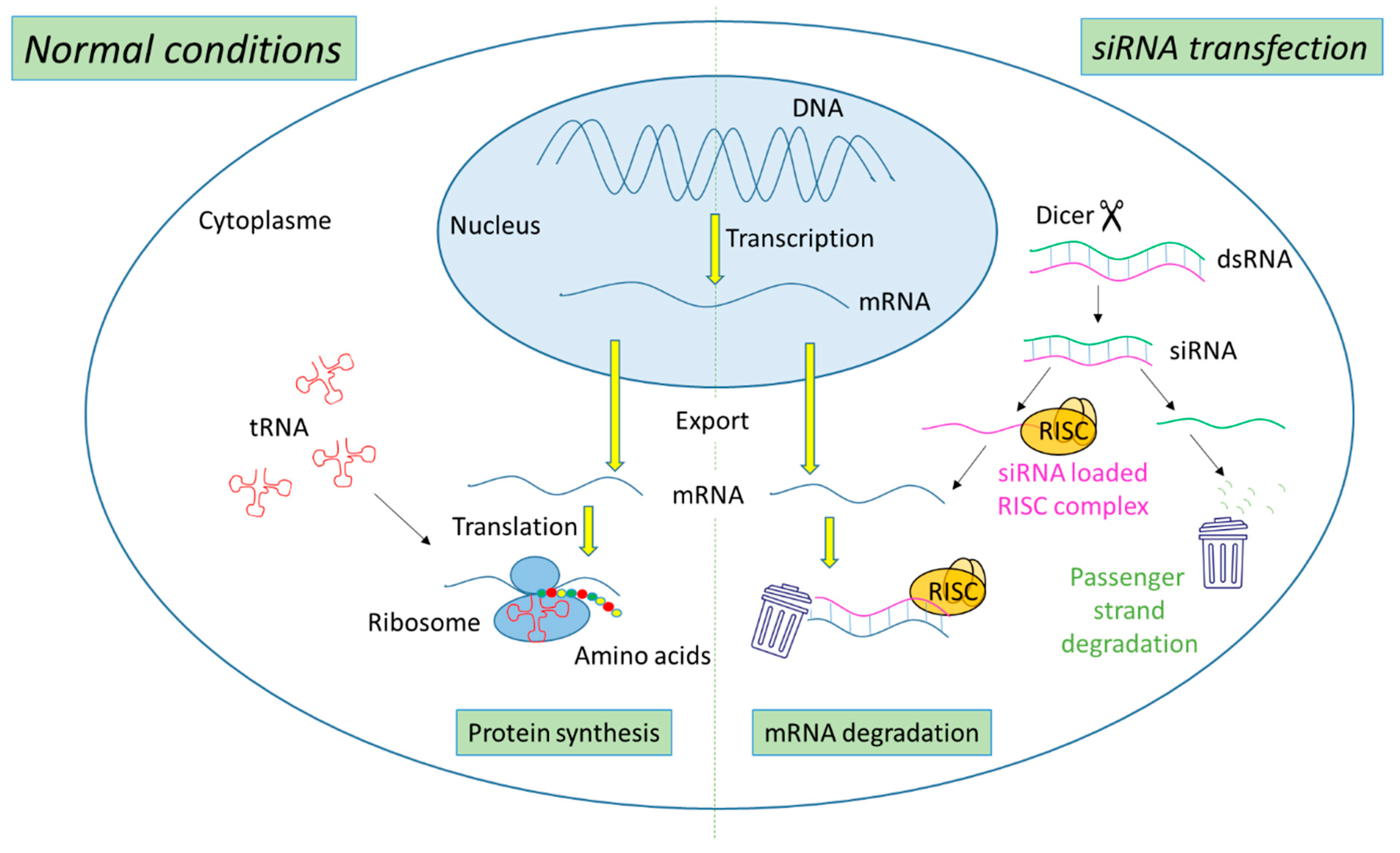
2.2. siRNA-Based Cancer Therapies
| Name/Sponsor | Route of Administration | Delivery System | Targeting Moiety | Target Gene | Disease | Clinical Trail Number (ClinicalTrials.gov) | Phase/Status | Period | Ref |
|---|---|---|---|---|---|---|---|---|---|
| CALAA-01/Calando Pharmaceuticals | i.v. | Cyclodextrin polymer-based nanoparticle | Transferrin | RRM2 | Solid tumors (Melanoma, gastrointestinal, prostate, etc.) | NCT00689065 | Phase I/Terminated | 2008–2012 | [21] |
| siG12D LODER/Silenseed Ltd. | Endoscopic intervention | Biodegradable Polymeric matrix | ----- | KRAS(G12D) and G12X mutations | Locally advanced pancreatic cancer | NCT01188785 | Phase I/Completed | 2011–2013 | [22] |
| siG12D-LODERs plus chemotherapy (Gemcitabine + nab-Paclitaxel or Folfirinox or modified Folfirinox) /Silenseed Ltd. | Endoscopic intervention | Biodegradable Polymeric matrix | ----- | KRAS(G12D) and G12X mutations | Locally advanced pancreatic cancer | NCT01676259 | Phase II/Recruiting | 2018–Est.2023 | [23] |
| ALN-VSP02/Alnylam Pharmaceuticals | i.v. | Lipid nanoparticle | ----- | VEGF KSP |
Solid tumors with liver involvement. | NCT00882180 NCT01158079 |
Phase I/Completed | 2009–2011 2010–2012 |
[24] |
| TKM-PLK1 (TKM-080301)/National Cancer Institute (NCI) | Hepatic Intra-Arterial Administration | Lipid nanoparticle | ----- | PLK1 | Primary or secondary liver cancer. | NCT01437007 | Phase I/Completed | 2011–2012 | [25] |
| Arbutus Biopharma Corporation | i.v. | Cancer, neuroendocrine tumors, adrenocortical carcinoma | NCT01262235 | Phase I/II/Completed | 2010–2015 | ||||
| Arbutus Biopharma Corporation | i.v. | Hepatocellular Carcinoma | NCT02191878 | Phase I/II/Completed | 2014–2016 | ||||
| DCR-MYC/Dicerna Pharmaceuticals, Inc. | i.v. | EnCoreTM lipid nanoparticle | ----- | MYC | Solid tumors, multiple myeloma, lymphoma | NCT02110563 | Phase I/Terminated | 2014–2016 | [26] |
| NBF-006/Nitto BioPharma, Inc. | Lipid nanoparticle | GSTP | Non-Small cell lung, pancreatic and colorectal Cancers | NCT03819387 | Phase I/Recruiting | 2019–Est.2023 | [27] | ||
| Atu027/Silence Therapeutics GmbH | i.v. | Liposomes | ----- | PKN3 | Advanced Solid Cancer | NCT00938574 | Phase I/Completed | 2009–2012 | [28] |
| Atu027-I-02 (Atu027 plus gemcitabine)/Silence Therapeutics GmbH | i.v. | Liposomes | ----- | PKN3 | Advanced or Metastatic Pancreatic Cancer | NCT01808638 | Phase I/II/Completed | 2013/2016 | [29] |
| EphA2-targeting DOPC-encapsulated siRNA/M.D. Anderson Cancer Center | i.v. | Liposomes | ----- | EphA2 | Advanced or recurrent solid tumors | NCT01591356 | Phase I/Active, not recruiting | 2015–Est.2024 | [30] |
| Mesenchymal Stromal Cells-derived Exosomes with KRAS(G12D) siRNA/M.D. Anderson Cancer Center | MSC exosome | CD47 | KRAS(G12D) | Metastatic pancreatic ductal adenocarcinoma with KrasG12D mutation | NCT03608631 | Phase I/Recruiting | 2021–Est.2023 | [31] |
2.3. Hurdles to siRNA Delivery
3. Photochemical Internalization (PCI) Mechanism
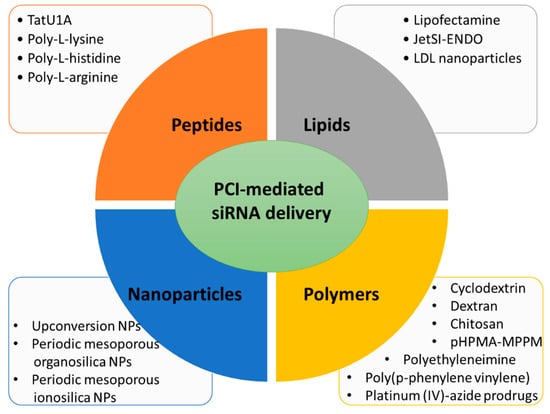
3.1. Lipid Carriers for PCI-Mediated siRNA Delivery
3.2. Peptide Carriers for PCI-Mediated siRNA Delivery
This entry is adapted from the peer-reviewed paper 10.3390/cancers14153597
References
- Yahya, E.B.; Alqadhi, A.M. Recent trends in cancer therapy: A review on the current state of gene delivery. Life Sci. 2021, 269, 119087.
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA A Cancer J. Clin. 2021, 71, 209–249.
- Kesse, S.; Boakye-Yiadom, K.O.; Ochete, B.O.; Opoku-Damoah, Y.; Akhtar, F.; Filli, M.S.; Farooq, M.A.; Aquib, M.; Mily, B.J.M.; Murtaza, G. Mesoporous Silica Nanomaterials: Versatile Nanocarriers for Cancer Theranostics and Drug and Gene Delivery. Pharmaceutics 2019, 11, 77.
- Ali, L.M.A. Toxicity Studies of Polymer Based Supermagnetic Iron Oxide Nanoparticles. Ph.D. Thesis, Universidad Zaragoza, Zaragoza, Spain, 2014.
- Duncan, R.; Gaspar, R. Nanomedicine(s) under the microscope. Mol. Pharm. 2011, 8, 2101–2141.
- Shi, J.; Kantoff, P.W.; Wooster, R.; Farokhzad, O.C. Cancer nanomedicine: Progress, challenges and opportunities. Nat. Rev. Cancer 2017, 17, 20–37.
- Mansoori, B.; Shotorbani, S.S.; Baradaran, B. RNA Interference and its Role in Cancer Therapy. Adv. Pharm. Bull. 2014, 4, 313–321.
- Fire, A.; Xu, S.; Montgomery, M.K.; Kostas, S.A.; Driver, S.E.; Mello, C.C. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 1998, 391, 806–811.
- Tan, F.L.; Yin, J.Q. RNAi, a new therapeutic strategy against viral infection. Cell Res. 2004, 14, 460–466.
- Holoch, D.; Moazed, D. RNA-mediated epigenetic regulation of gene expression. Nat. Rev. Genet. 2015, 16, 71–84.
- Hammond, S.M.; Caudy, A.A.; Hannon, G.J. Post-transcriptional gene silencing by double-stranded RNA. Nat. Rev. Genet. 2001, 2, 110–119.
- Yoon, J.; Shin, M.; Lee, J.Y.; Lee, S.N.; Choi, J.H.; Choi, J.W. RNA interference (RNAi)-based plasmonic nanomaterials for cancer diagnosis and therapy. J. Control. Release 2022, 342, 228–240.
- Pai, S.; Lin, Y.Y.; Macaes, B.; Meneshian, A.; Hung, C.F.; Wu, T.C. Prospects of RNA interference therapy for cancer. Gene Ther. 2006, 13, 464–477.
- Pengnam, S.; Plianwong, S.; Yingyongnarongkul, B.-k.; Patrojanasophon, P.; Opanasopit, P. Delivery of small interfering RNAs by nanovesicles for cancer therapy. Drug Metab. Pharmacokinet. 2022, 42, 100425.
- Miele, E.; Spinelli, G.P.; Miele, E.; Di Fabrizio, E.; Ferretti, E.; Tomao, S.; Gulino, A. Nanoparticle-based delivery of small interfering RNA: Challenges for cancer therapy. Int. J. Nanomed. 2012, 7, 3637–3657.
- Kim, D.H.; Rossi, J.J. Strategies for silencing human disease using RNA interference. Nat. Rev. Genet. 2007, 8, 173–184.
- Lares, M.R.; Rossi, J.J.; Ouellet, D.L. RNAi and small interfering RNAs in human disease therapeutic applications. Trends Biotechnol. 2010, 28, 570–579.
- de Fougerolles, A.; Vornlocher, H.P.; Maraganore, J.; Lieberman, J. Interfering with disease: A progress report on siRNA-based therapeutics. Nat. Rev. Drug Discov. 2007, 6, 443–453.
- Dönmez, Y.; Gündüz, U. Reversal of multidrug resistance by small interfering RNA (siRNA) in doxorubicin-resistant MCF-7 breast cancer cells. Biomed. Pharmacother. 2011, 65, 85–89.
- Singh, A.; Trivedi, P.; Jain, N.K. Advances in siRNA delivery in cancer therapy. Artif. Cells Nanomed. Biotechnol. 2017, 46, 274–283.
- Zuckerman, J.E.; Gritli, I.; Tolcher, A.; Heidel, J.D.; Lim, D.; Morgan, R.; Chmielowski, B.; Ribas, A.; Davis, M.E.; Yen, Y. Correlating animal and human phase Ia/Ib clinical data with CALAA-01, a targeted, polymer-based nanoparticle containing siRNA. Proc. Natl. Acad. Sci. USA 2014, 111, 11449–11454.
- Golan, T.; Khvalevsky, E.Z.; Hubert, A.; Gabai, R.M.; Hen, N.; Segal, A.; Domb, A.; Harari, G.; Ben David, E.; Raskin, S.; et al. RNAi therapy targeting KRAS in combination with chemotherapy for locally advanced pancreatic cancer patients. Oncotarget 2015, 6, 24560–24570.
- Varghese, A.M.; Ang, C.; DiMaio, C.J.; Javle, M.M.; Gutierrez, M.; Yarom, N.; Stemmer, S.M.; Golan, T.; Geva, R.; Semenisty, V.; et al. A phase II study of siG12D-LODER in combination with chemotherapy in patients with locally advanced pancreatic cancer (PROTACT). J. Clin. Oncol. 2020, 38, TPS4672.
- Tabernero, J.; Shapiro, G.I.; Lorusso, P.M.; Cervantes, A.; Schwartz, G.K.; Weiss, G.J.; Paz-Ares, L.; Cho, D.C.; Infante, J.R.; Alsina, M.; et al. First-in-Humans Trial of an RNA Interference Therapeutic Targeting VEGF and KSP in Cancer Patients with Liver Involvement. Cancer Discov. 2013, 3, 406–417.
- Barba, A.A.; Bochicchio, S.; Dalmoro, A.; Lamberti, G. Lipid Delivery Systems for Nucleic-Acid-Based-Drugs: From Production to Clinical Applications. Pharmaceutics 2019, 11, 360.
- Tolcher, A.W.; Papadopoulos, K.P.; Patnaik, A.; Rasco, D.W.; Martinez, D.; Wood, D.L.; Fielman, B.; Sharma, M.; Janisch, L.A.; Brown, B.D.; et al. Safety and activity of DCR-MYC, a first-in-class Dicer-substrate small interfering RNA (DsiRNA) targeting MYC, in a phase I study in patients with advanced solid tumors. J. Clin. Oncol. 2015, 33, 11006.
- A Study of NBF-006 in Non-Small Cell Lung, Pancreatic, or Colorectal Cancer. Available online: https://clinicaltrials.gov/ct2/show/NCT03819387 (accessed on 3 May 2022).
- Schultheis, B.; Strumberg, D.; Santel, A.; Vank, C.; Gebhardt, F.; Keil, O.; Lange, C.; Giese, K.; Kaufmann, J.; Khan, M.; et al. First-in-Human Phase I Study of the Liposomal RNA Interference Therapeutic Atu027 in Patients With Advanced Solid Tumors. J. Clin. Oncol. 2014, 32, 4141–4148.
- Schultheis, B.; Strumberg, D.; Kuhlmann, J.; Wolf, M.; Link, K.; Seufferlein, T.; Kaufmann, J.; Feist, M.; Gebhardt, F.; Khan, M.; et al. Safety, Efficacy and Pharcacokinetics of Targeted Therapy with The Liposomal RNA Interference Therapeutic Atu027 Combined with Gemcitabine in Patients with Pancreatic Adenocarcinoma. A Randomized Phase Ib/IIa Study. Cancers 2020, 12, 3130.
- Oner, E.; Kotmakci, M.; Baird, A.M.; Gray, S.G.; Butuner, B.D.; Bozkurt, E.; Kantarci, A.G.; Finn, S.P. Development of EphA2 siRNA-loaded lipid nanoparticles and combination with a small-molecule histone demethylase inhibitor in prostate cancer cells and tumor spheroids. J. Nanobiotechnol. 2021, 19, 71.
- Lee, B.-C.; Kang, I.; Yu, K.-R. Therapeutic Features and Updated Clinical Trials of Mesenchymal Stem Cell (MSC)-Derived Exosomes. J. Clin. Med. 2021, 10, 711.
- Zhang, M.M.; Bahal, R.; Rasmussen, T.P.; Manautou, J.E.; Zhong, X.-B. The growth of siRNA-based therapeutics: Updated clinical studies. Biochem. Pharmacol. 2021, 189, 114432.
- Liu, F.; Shollenberger, L.; Conwell, C.C.; Yuan, X.; Huang, L. Mechanism of naked DNA clearance after intravenous injection. J. Gene Med. 2007, 9, 613–619.
- Kanasty, R.L.; A Whitehead, K.; Vegas, A.J.; Anderson, D.G. Action and Reaction: The Biological Response to siRNA and Its Delivery Vehicles. Mol. Ther. 2012, 20, 513–524.
- Sharma, V.K.; Sharma, R.K.; Singh, S.K. Antisense oligonucleotides: Modifications and clinical trials. MedChemComm 2014, 5, 1454–1471.
- Nair, J.K.; Willoughby, J.L.S.; Chan, A.; Charisse, K.; Alam, R.; Wang, Q.; Hoekstra, M.; Kandasamy, P.; Kel’In, A.V.; Milstein, S.; et al. Multivalent N-Acetylgalactosamine-Conjugated siRNA Localizes in Hepatocytes and Elicits Robust RNAi-Mediated Gene Silencing. J. Am. Chem. Soc. 2014, 136, 16958–16961.
- Gangopadhyay, S.; Gore, K.R. Advances in siRNA therapeutics and synergistic effect on siRNA activity using emerging dual ribose modifications. RNA Biol. 2022, 19, 452–467.
- Thomas, C.E.; Ehrhardt, A.; Kay, M.A. Progress and problems with the use of viral vectors for gene therapy. Nat. Rev. Genet. 2003, 4, 346–358.
- Mikami, A.; Erande, N.; Matsuda, S.; Kel’in, A.; Woods, L.B.; Chickering, T.; Pallan, P.S.; Schlegel, M.K.; Zlatev, I.; Egli, M.; et al. Synthesis, chirality-dependent conformational and biological properties of siRNAs containing 5′-(R)- and 5′-(S)-C-methyl-guanosine. Nucleic Acids Res. 2020, 48, 10101–10124.
- Nel, A.E.; Mädler, L.; Velegol, D.; Xia, T.; Hoek, E.M.V.; Somasundaran, P.; Klaessig, F.; Castranova, V.; Thompson, M. Understanding biophysicochemical interactions at the nano–bio interface. Nat. Mater. 2009, 8, 543–557.
- Guo, S.; Huang, L. Nanoparticles Escaping RES and Endosome: Challenges for siRNA Delivery for Cancer Therapy. J. Nanomater. 2011, 2011, 1–12.
- Yezhelyev, M.V.; Qi, L.; O’Regan, R.M.; Nie, S.; Gao, X. Proton-Sponge Coated Quantum Dots for siRNA Delivery and Intracellular Imaging. J. Am. Chem. Soc. 2008, 130, 9006–9012.
- Hoffmann, M.; Hersch, N.; Merkel, R.; Csiszar, A.; Hoffmann, B. Changing the Way of Entrance: Highly Efficient Transfer of mRNA and siRNA via Fusogenic Nano-Carriers. J. Biomed. Nanotechnol. 2019, 15, 170–183.
- Otterhaug, T.; Janetzki, S.; Welters, M.J.P.; Håkerud, M.; Nedberg, A.G.; Edwards, V.T.; Boekestijn, S.; Loof, N.M.; Selbo, P.K.; Olivecrona, H.; et al. Photochemical Internalization Enhanced Vaccination Is Safe, and Gives Promising Cellular Immune Responses to an HPV Peptide-Based Vaccine in a Phase I Clinical Study in Healthy Volunteers. Front. Immunol. 2021, 11, 576756.
- Dechêne, A.; Kasper, S.; Olivecrona, H.; Schirra, J.; Trojan, J. Photochemical internalization and gemcitabine combined with first-line chemotherapy in perihilar cholangiocarcinoma: Observations in three patients. Endosc. Int. Open 2020, 8, E1878–E1883.
- Ożog, L.; Aebisher, D. Singlet oxygen lifetime and diffusion measurements. Eur. J. Clin. Exp. Med. 2018, 16, 123–126.
- Klaper, M.; Fudickar, W.; Linker, T. Role of Distance in Singlet Oxygen Applications: A Model System. J. Am. Chem. Soc. 2016, 138, 7024–7029.
- Raemdonck, K.; Naeye, B.; Høgset, A.; Demeester, J.; De Smedt, S.C. Prolonged gene silencing by combining siRNA nanogels and photochemical internalization. J. Control. Release 2010, 145, 281–288.
- Berg, K.; Folini, M.; Prasmickaite, L.; Selbo, P.; Bonsted, A.; Engesaeter, B.; Zaffaroni, N.; Weyergang, A.; Dietzea, A.; Maelandsmo, G.; et al. Photochemical Internalization: A New Tool for Drug Delivery. Curr. Pharm. Biotechnol. 2007, 8, 362–372.
- Berg, K.; Selbo, P.K.; Prasmickaite, L.; E Tjelle, T.; Sandvig, K.; Moan, J.; Gaudernack, G.; Fodstad, O.; Kjølsrud, S.; Anholt, H.; et al. Photochemical internalization: A novel technology for delivery of macromolecules into cytosol. Cancer Res. 1999, 59, 1180–1183.
- Selbo, P.K.; Sivam, G.; Fodstad, O.; Sandvig, K.; Berg, K. In vivo documentation of photochemical internalization, a novel approach to site specific cancer therapy. Int. J. Cancer 2001, 92, 761–766.
- Berg, K.; Dietze, A.; Kaalhus, O.; Høgset, A. Site-Specific Drug Delivery by Photochemical Internalization Enhances the Antitumor Effect of Bleomycin. Clin. Cancer Res. 2005, 11, 8476–8485.
- Sultan, A.; Jerjes, W.; Berg, K.; Høgset, A.; Mosse, C.A.; Hamoudi, R.; Hamdoon, Z.; Simeon, C.; Carnell, D.; Forster, M.; et al. Disulfonated tetraphenyl chlorin (TPCS2a)-induced photochemical internalisation of bleomycin in patients with solid malignancies: A phase 1, dose-escalation, first-in-man trial. Lancet Oncol. 2016, 17, 1217–1229.
- Oliveira, S.; Fretz, M.M.; Høgset, A.; Storm, G.; Schiffelers, R.M. Photochemical internalization enhances silencing of epidermal growth factor receptor through improved endosomal escape of siRNA. Biochim. et Biophys. Acta (BBA)-Biomembr. 2007, 1768, 1211–1217.
- Kraja, I.; Bing, R.; Hiwatashi, N.; Rousseau, B.; Nalband, D.; Kirshenbaum, K.; Branski, R.C. Preliminary study of a novel transfection modality for in vivo siRNA delivery to vocal fold fibroblasts. Laryngoscope 2017, 127, E231–E237.
- Bøe, S.; Longva, A.; Hovig, E. Photochemically Induced Gene Silencing Using Small Interfering RNA Molecules in Combination with Lipid Carriers. Oligonucleotides 2007, 17, 166–173.
- Jin, H.; Lovell, J.F.; Chen, J.; Lin, Q.; Ding, L.; Ng, K.K.; Pandey, R.K.; Manoharan, M.; Zhang, Z.; Zheng, G. Mechanistic Insights into LDL Nanoparticle-Mediated siRNA Delivery. Bioconjug. Chem. 2011, 23, 33–41.
- Schwarze, S.R.; Ho, A.; Vocero-Akbani, A.; Dowdy, S.F. In Vivo Protein Transduction: Delivery of a Biologically Active Protein into the Mouse. Science 1999, 285, 1569–1572.
- Del Gaizo, V.; Payne, R.M. A novel TAT-mitochondrial signal sequence fusion protein is processed, stays in mitochondria, and crosses the placenta. Mol. Ther. 2003, 7, 720–730.
- Endoh, T.; Sisido, M.; Ohtsuki, T. Cellular siRNA Delivery Mediated by a Cell-Permeant RNA-Binding Protein and Photoinduced RNA Interference. Bioconjug. Chem. 2008, 19, 1017–1024.
- Maiolo, J.R.; Ottinger, E.A.; Ferrer, M. Specific Redistribution of Cell-Penetrating Peptides from Endosomes to the Cytoplasm and Nucleus upon Laser Illumination. J. Am. Chem. Soc. 2004, 126, 15376–15377.
- Matsushita, M.; Noguchi, H.; Lu, Y.-F.; Tomizawa, K.; Michiue, H.; Li, S.-T.; Hirose, K.; Bonner-Weir, S.; Matsui, H. Photo-acceleration of protein release from endosome in the protein transduction system. FEBS Lett. 2004, 572, 221–226.
- Jørgensen, J.A.L.; Longva, A.S.; Hovig, E.; Bøe, S.L. Evaluation of Biodegradable Peptide Carriers for Light-Directed Targeting. Nucleic Acid Ther. 2013, 23, 131–139.
- Soe, T.H.; Watanabe, K.; Ohtsuki, T. Photoinduced Endosomal Escape Mechanism: A View from Photochemical Internalization Mediated by CPP-Photosensitizer Conjugates. Molecules 2020, 26, 36.
- Wang, J.T.-W.; Giuntini, F.; Eggleston, I.M.; Bown, S.G.; MacRobert, A.J. Photochemical internalisation of a macromolecular protein toxin using a cell penetrating peptide-photosensitiser conjugate. J. Control. Release 2012, 157, 305–313.
- Ohtsuki, T.; Miki, S.; Kobayashi, S.; Haraguchi, T.; Nakata, E.; Hirakawa, K.; Sumita, K.; Watanabe, K.; Okazaki, S. The molecular mechanism of photochemical internalization of cell penetrating peptide-cargo-photosensitizer conjugates. Sci. Rep. 2015, 5, 18577.
- Sarko, D.; Beijer, B.; Garcia Boy, R.; Nothelfer, E.-M.; Leotta, K.; Eisenhut, M.; Altmann, A.; Haberkorn, U.; Mier, W. The Pharmacokinetics of Cell-Penetrating Peptides. Mol. Pharm. 2010, 7, 2224–2231.
- Krämer, S.; Wunderli-Allenspach, H. No entry for TAT(44–57) into liposomes and intact MDCK cells: Novel approach to study membrane permeation of cell-penetrating peptides. Biochim. et Biophys. Acta (BBA)-Biomembr. 2002, 1609, 161–169.
- Mitchell, D.; Steinman, L.; Kim, D.; Fathman, C.; Rothbard, J. Polyarginine enters cells more efficiently than other polycationic homopolymers. J. Pept. Res. 2000, 56, 318–325.