 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Gaetana Paolella | -- | 3687 | 2022-07-18 17:57:20 | | | |
| 2 | Sirius Huang | + 2 word(s) | 3689 | 2022-07-22 03:01:58 | | | | |
| 3 | Sirius Huang | Meta information modification | 3689 | 2022-07-22 03:03:28 | | |
Video Upload Options
Coeliac disease (CD) is a multifactiorial enteropathy that affects the small intestine of genetically predisposed individuals. A condition of partial to total atrophy, together with crypt hyperplasia and consistent lymphocytic infiltration, characterises the intestinal mucosa of affected patients. The main environmental trigger is a heterogenic proteic component of some dietary cereals, commonly known as gluten. A strong immune response against gluten, both cellular and humoral, is mounted in CD, accompanied by a humoral autoimmune response against self-proteins, in particular type 2 transglutaminase (TG2).
1. Epidemiology and Clinical Manifestations of CD
2. The Main Environmental Trigger in CD: Gluten
3. The Genetics of CD
4. The Adaptive and Innate Immune Response in CD
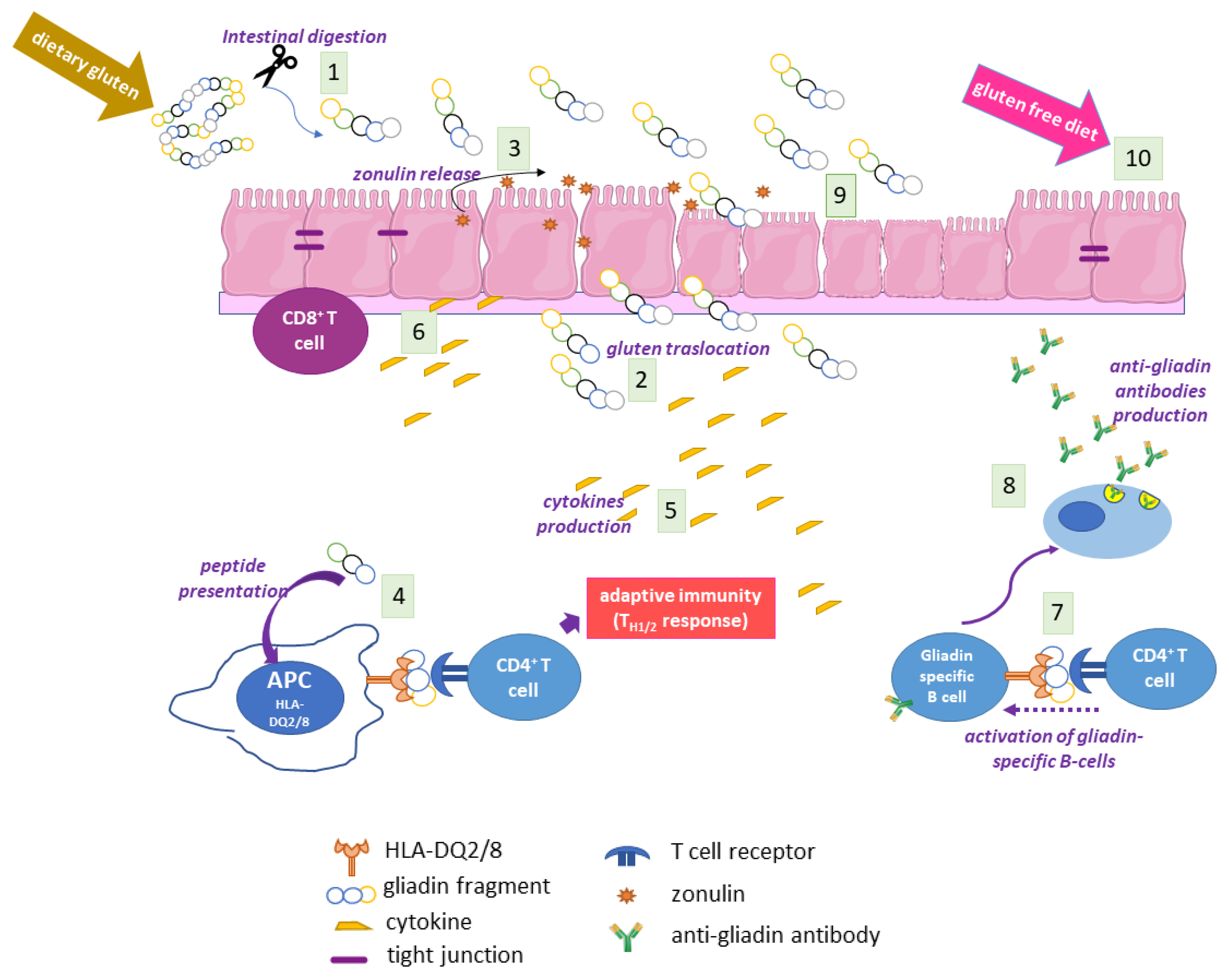
5. Autoimmunity in CD
6. The Coeliac Cellular Phenotype
7. CD Diagnosis
Enzymatic and Immunofluorescence Assays
8. CD Therapy
9. TG2
The TG family includes transferase enzymes (EC 2.3.2.13), which catalyse the formation of an isopeptide bond involving the γ-carboxamide group of a Gln residue (the acyl-donor) and the ε-amino group of a Lys residue (the nucleophilic acyl-acceptor), generating an intra- or an inter-protein cross-link [36]. The biochemical mechanism of the reaction involves a ping-pong kinetics: first, the γ-carboxamide group forms a thiol ester with the Cys residue of the enzyme active site with the release of ammonia, then the acyl intermediate is transferred to the nucleophilic aminic substrate. As alternatives to Lys residues as acyl-acceptors in the crosslinking reaction, TGs can use several low-molecular-mass primary amines (spermine, spermidine, putrescine, etc.). In the absence of suitable amines and in the presence of a slightly acidic pH, water can act as nucleophile; thus, TGs are able to deamidate Gln to Glu residues [36]. In mammals, nine structurally and functionally related genes encoding TGs have been identified [37]. Among them, only one gene encodes for a protein (band 4.2) without catalytic activity. All other eight mammalian TGs are strictly Ca2+-dependent enzymes and share a high sequence identity around the Cys proteinase-active site, comprising the catalytic triad Cys, His, and Asp [36]. Moreover, the structural organisation in four sequential domains is highly conserved in TG isoforms: the N-terminal β-sandwich domain is followed by the catalytic core domain (containing the Ca2+ binding sites) and two C-terminal β-barrel domains [36].
10. TG2 in CD Pathogenesis
10.1. Gluten Modification: Deamidation
Given the high content of Gln residues in gliadin and other gluten proteins, it is evident that TG2 can use them as Gln-donor substrates in transamidating and deamidating reactions. Deamidation is not a common reaction because it requires specific conditions: a slightly acidic pH and a very low concentration of available amines. When and where these conditions may be realised in cells and tissues are still debated. In a recent work, it has been suggested that TG2 released in gut lumen as a consequence of enterocytes shedding could meet luminal gliadin, and in this way could have direct access to adaptive immune B cells at the level of Peyer’s patches [42]. TG2 on the cell surfaces of APCs could deamidate gliadin during its turnover by endocytosis at the level of acidic endocytic vesicles. The recent demonstration that TG2 is more abundant on the cell surface and in early endocytic vesicles of CD skin-derived fibroblasts than of control cells supports this hypothesis [24]. In any case, there is no doubt that TG2 recognises specific Gln residues in gliadin sequences and deamidates them, introducing a net negative charge that greatly increases the affinity with which DQ2/DQ8 heterodimers bind gliadin peptides for presentation to T cells [43]. Indeed, the production of high-resolution X-ray crystal structures of representative deamidated gluten peptides in complex with DQ2 and DQ8 has definitively confirmed the importance of the introduction of a negative charge at a specific location in a gluten peptide interacting with the HLA binding groove [44]. As a consequence, a stronger immune response is evoked. An updated list of CD-relevant deamidated gluten epitopes recognised by CD4+ T cells has recently been reported [5]. The majority of listed epitopes are DQ2.5-restricted and mainly belong to the α- and γ-gliadins, but some epitopes also belong to the ϖ-gliadins, glutenins, hordeins, secalins, and avenins. Only a few epitopes are DQ2.2-or DQ8-restricted and also belong to the α- and γ-gliadins and glutenins. This discrepancy is likely due to the fact that most gluten epitopes have been discovered in CD patients positive for HLA-DQ2.5 [5]. Thus, it is reasonable to expect that further additional epitopes will be defined in the future.
10.2. Gluten Modification: Mechanism of Anti-TG2 Antibody Production
11. Conclusion
It is doubtless that TG2 represents a key enzyme in the pathogenic mechanisms leading to CD development. At the site of intestinal inflammation, where TG2 is typically overexpressed, its canonical enzymatic activity leads to the formation of isopeptide bonds between itself (or other self-proteins) and gluten peptides, thus provoking a strong autoimmune response. Still uncertain is the role of circulating and intestinal anti-TG2 antibodies in contributing to CD onset; however, antibody detection represents an irreplaceable tool for CD screening programs and diagnosis. In particular circumstances (low pH and virtual absence of primary amines) in the CD mucosa, TG2 can modify specific Gln residues of gluten peptides, transforming a neutral residue into an acidic residue (Glu), which is important in the presentation to the immune system by DQ2/8 molecules and the consequent loss of oral tolerance towards gluten. For these evident reasons, TG2 has become the targetof recent pharmaceutical approaches aimed at reducing deamidation. New generations of increasingly specific and potent TG2 inhibitors are under investigation, some of which have reached the point of clinical experimentation. Finally, given the increasing use of food-grade TGs, particularly TGm, as biotechnological tools in a number of industrial applications, some researchers are exploring the possibility of modifying gluten peptides by transamidating them with nucleophilic amines in an attempt to reduce the immunogenicity of the majority of DQ2/8-restricted epitopes. Finally, it is appropriate to underline that TG2 is a multifunctional enzyme with several catalytic activities and non-catalytic functions. Thus, it cannot be excluded that TG2 may have other unexpected roles in CD pathogenesis, independent of its transamidating and deamidating activity. For example, TG2 is easily targeted on the cell surface by anti-TG2 antibodies, and not only at the intestinal level. Given the biological consequences of such an interaction at the cell surface (for example, increased proliferation and structural and signalling modifications) and potential differences in subcellular TG2 distribution in CD cells, it is reasonable to conclude that TG2 may contribute to CD onset and progression in other peculiar and still unexplored fashions.
References
- Caio, C.; Volta, U.; Sapone, A.; Leffler, D.A.; De Giorgio, R.; Catassi, C.; Alessio, F. Celiac disease: A comprehensive current review. BMC Med. 2019, 17, 142.
- Durazzo, M.; Ferro, A.; Brascugli, I.; Mattivi, S.; Fagoonee, S.; Pellicano, R. Extra-Intestinal Manifestations of Celiac Disease: What Should We Know in 2022? J. Clin. Med. 2022, 11, 258.
- Wieser, H. Chemistry of gluten proteins. Food Microbiol. 2007, 24, 115–119.
- Daly, M.; Bromilow, S.N.; Nitride, C.; Shewry, P.R.; Gethings, L.A.; Mills, E.N.C. Mapping Coeliac Toxic Motifs in the Prolamin Seed Storage Proteins of Barley, Rye, and Oats Using a Curated Sequence Database. Front. Nutr. 2020, 7, 87.
- Sollid, L.M.; Tye-Din, J.A.; Qiao, S.W.; Anderson, R.P.; Gianfrani, C.; Koning, F. Update 2020: Nomenclature and listing of celiac disease-relevant gluten epitopes recognized by CD4(+) T cells. Immunogenetics 2020, 72, 85–88.
- Shan, L.; Molberg, Ø.; Parrot, I.; Hausch, F.; Filiz, F.; Gray, G.M.; Sollid, L.M.; Khosla, C. Structural basis for gluten intolerance in celiac sprue. Science 2002, 297, 2275–2279.
- Sollid, L.M.; Jabri, B. Celiac disease and transglutaminase 2: A model for posttranslational modification of antigens and HLA association in the pathogenesis of auto-immune disorders. Curr. Opin. Immunol. 2011, 23, 732–738.
- Sollid, L.M. Molecular basis of celiac disease. Annu. Rev. Immunol. 2000, 18, 53–81.
- Voisine, J.; Abadie, V. Interplay Between Gluten, HLA, Innate and Adaptive Immunity Orchestrates the Development of Coeliac Disease. Front. Immunol. 2021, 12, 674313.
- Bodkhe, R.; Shetty, S.A.; Dhotre, D.P.; Verma, A.K.; Bhatia, K.; Mishra, A.; Kaur, G.; Pande, P.; Bangarusamy, D.K.; Santosh, B.P.; et al. Comparison of small gut and whole gut microbiota of first-degree relatives with adult celiac disease patients and controls. Front. Microbiol. 2019, 10, 137–140.
- Kemppainen, K.M.; Lynch, K.F.; Liu, E.; Lönnrot, M.; Simell, V.; Briese, T.; Koletzko, S.; Hagopian, W.; Rewers, M.; She, J.X.; et al. Factors that increase risk of celiac disease autoimmunity after a gastrointestinal infection in early life. Clin. Gastroenterol. Hepatol. 2017, 15, 694–702.e5.
- Sollid, L.M. The roles of MHC class II genes and post-translational modification in celiac disease. Immunogenetics 2017, 69, 605–616.
- Kårhus, L.L.; Thuesen, B.H.; Skaaby, T.; Rumessen, J.J.; Linneberg, A. The distribution of HLA DQ2 and DQ8 haplotypes and their association with health indicators in a general Danish population. United Eur. Gastroenterol. J. 2018, 6, 866–878.
- García-Santisteban, I.; Romero-Garmendia, I.; Cilleros-Portet, A.; Bilbao, J.R.; Fernandez-Jimenez, N. Celiac disease susceptibility: The genome and beyond. Int. Rev. Cell. Mol. Biol. 2021, 358, 1–45.
- Lammers, K.M.; Lu, R.; Brownley, J.; Lu, B.; Gerard, C.; Thomas, K.; Rallabhandi, P.; Shea-Donohue, T.; Tamiz, A.; Alkan, S.; et al. Gliadin induces an increase in intestinal permeability and zonulin release by binding to the chemokine receptor CXCR3. Gastroenterology 2008, 135, 194–204.e3.
- Chirdo, F.G.; Auricchio, S.; Troncone, R.; Barone, M.V. The gliadin p31-43 peptide: Inducer of multiple proinflammatory effects. Int. Rev. Cell. Mol. Biol. 2021, 358, 165–205.
- Dieterich, W.; Ehnis, T.; Bauer, M.; Donner, P.; Volta, U.; Riecken, E.O.; Schuppan, D. Identification of tissue transglutaminase as the autoantigen of celiac disease. Nat. Med. 1997, 3, 797–801.
- Kaukinen, K.; Peräaho, M.; Collin, P.; Partanen, J.; Woolley, N.; Kaartinen, T.; Nuutinen, T.; Halttunen, T.; Mäki, M.; Korponay-Szabo, I. Small-bowel mucosal transglutaminase 2-specific IgA deposits in coeliac disease without villous atrophy: A prospective and randomized clinical study. Scand. J. Gastroenterol. 2005, 40, 564–572.
- Korponay-Szabó, I.R.; Halttunen, T.; Szalai, Z.; Laurila, K.; Király, R.; Kovács, J.B.; Fésüs, L.; Mäki, M. In vivo targeting of intestinal and extraintestinal transglutaminase 2 by coeliac auto-antibodies. Gut 2004, 53, 641–648.
- Sardy, M.; Karpati, S.; Merkl, B.; Paulsson, M.; Smyth, N. Epidermal transglutaminase (TGase 3) is the autoantigen of dermatitis herpetiformis. J. Exp. Med. 2002, 195, 747–757.
- Hadjivassiliou, M.; Aeschlimann, P.; Strigun, A.; Sanders, D.S.; Woodroofe, N.; Aeschlimann, D. Auto-antibodies in gluten ataxia recognize a novel neuronal transglutaminase. Ann. Neurol. 2008, 64, 332–343.
- Nanayakkara, M.; Lania, G.; Maglio, M.; Kosova, R.; Sarno, M.; Gaito, A.; Discepolo, V.; Troncone, R.; Auricchio, S.; Auricchio, R.; et al. Enterocyte proliferation and signaling are constitutively altered in celiac disease. PLoS ONE 2013, 8, e76006.
- Lania, G.; Nanayakkara, M.; Maglio, M.; Auricchio, R.; Porpora, M.; Conte, M.; De Matteis, M.M.; Rizzo, R.; Luini, A.; Discepolo, V.; et al. Constitutive alterations in vesicular trafficking increase the sensitivity of cells from celiac disease patients to gliadin. Commun. Biol. 2019, 2, 190.
- Paolella, G.; Nanayakkara, M.; Sposito, S.; Lepretti, M.; Auricchio, S.; Esposito, C.; Barone, M.V.; Martucciello, S.; Caputo, I. Constitutive differential features of type 2 transglutaminase in cells derived from celiac patients and from healthy subjects. Int. J. Mol. Sci. 2020, 21, 1231.
- Discepolo, V.; Lania, G.; Ten Eikelder, M.L.G.; Nanayakkara, M.; Sepe, L.; Tufano, R.; Troncone, R.; Auricchio, S.; Auricchio, R.; Paolella, G.; et al. Pediatric Celiac Disease Patients Show Alterations of Dendritic Cell Shape and Actin Rearrangement. Int. J. Mol. Sci. 2021, 22, 2708.
- Porpora, M.; Conte, M.; Lania, G.; Bellomo, C.; Rapacciuolo, L.; Chirdo, F.G.; Auricchio, R.; Troncone, R.; Auricchio, S.; Barone, M.V.; et al. Inflammation Is Present, Persistent and More Sensitive to Proinflammatory Triggers in Celiac Disease Enterocytes. Int. J. Mol. Sci. 2022, 23, 1973.
- Gandini, A.; Gededzha, M.P.; De Maayer, T.; Barrow, P.; Mayne, E. Diagnosing coeliac disease: A literature review. Hum. Immunol. 2021, 82, 930–936.
- Tan, I.L.; Coutinho de Almeida, R.; Modderman, R.; Stachurska, A.; Dekens, J.; Barisani, D.; Meijer, C.R.; Roca, M.; Martinez-Ojinaga, E.; Shamir, R.; et al. Circulating miRNAs as Potential Biomarkers for Celiac Disease Development. Front. Immunol. 2021, 12, 734763.
- Volta, U.; Fabbri, A.; Parisi, C.; Piscaglia, M.; Caio, G.; Tovoli, F.; Fiorini, E. Old and new serological tests for celiac disease screening. Expert Rev. Gastroenterol. Hepatol. 2010, 4, 31–35.
- Rauhavirta, T.; Hietikko, M.; Salmi, T.; Lindfors, K. Transglutaminase 2 and Transglutaminase 2 Autoantibodies in Celiac Disease: A Review. Clin. Rev. Allergy Immunol. 2019, 57, 23–38.
- Choung, R.S.; Khaleghi Rostamkolaei, S.; Ju, J.M.; Marietta, E.V.; Van Dyke, C.T.; Rajasekaran, J.J.; Jayaraman, V.; Wang, T.; Bei, K.; Rajasekaran, K.E.; et al. Synthetic Neoepitopes of the Transglutaminase-Deamidated Gliadin Complex as Biomarkers for Diagnosing and Monitoring Celiac Disease. Gastroenterology 2019, 156, 582–591.e1.
- Ladinser, B.; Rossipal, E.; Pittschieler, K. Endomysium antibodies in coeliac disease: An improved method. Gut 1994, 35, 776–778.
- Balakireva, A.V.; Zamyatnin, A.A. Properties of Gluten Intolerance: Gluten Structure, Evolution, Pathogenicity and Detoxification Capabilities. Nutrients 2016, 8, 644.
- Chetcuti Zammit, S.; Sanders, D.S.; Sidhu, R. Refractory coeliac disease: What should we be doing different? Curr. Opin. Gastroenterol. 2020, 36, 215–222.
- Klonarakis, M.; Andrews, C.N.; Raman, M.; Panaccione, R.; Ma, C. Review article: Therapeutic targets for the pharmacologic management of coeliac disease-the future beyond a gluten-free diet. Aliment. Pharmacol. Ther. 2022, 55, 1277–1296.
- Lorand, L.; Graham, R.M. Transglutaminases: Crosslinking enzymes with pleiotropic functions. Nat. Rev. Mol. Cell. Biol. 2003, 4, 140–156.
- Grenard, P.; Bates, M.K.; Aeschlimann, D. Evolution of transglutaminase genes: Identification of a transglutaminase gene cluster on human chromosome 15q15. Structure of the gene encoding transglutaminase X and a novel gene family member, transglutaminase Z. J. Biol. Chem. 2001, 276, 33066–33078.
- Szondy, Z.; Korponay-Szabó, I.; Király, R.; Sarang, Z.; Tsay, G.J. Transglutaminase 2 in human diseases. Biomedicine 2017, 7, 15.
- Tempest, R.; Guarnerio, S.; Maani, R.; Cooper, J.; Peake, N. The Biological and Biomechanical Role of Transglutaminase-2 in the Tumour Microenvironment. Cancers 2021, 13, 2788.
- Chen, S.; Ma, J.; Chi, J.; Zhang, B.; Zheng, X.; Chen, J.; Liu, J. Roles and potential clinical implications of tissue transglutaminase in cardiovascular diseases. Pharmacol. Res. 2022, 177, 106085.
- Martucciello, S.; Sposito, S.; Esposito, C.; Paolella, G.; Caputo, I. Interplay between Type 2 Transglutaminase (TG2), Gliadin Peptide 31-43 and Anti-TG2 Antibodies in Celiac Disease. Int. J. Mol. Sci. 2020, 21, 3673.
- Iversen, R.; Amundsen, S.F.; Kleppa, L.; du Pré, M.F.; Stamnaes, J.; Sollid, L.M. Evidence That Pathogenic Transglutaminase 2 in Celiac Disease Derives from Enterocytes. Gastroenterology 2020, 159, 788–790.
- Ciacchi, L.; Reid, H.H.; Rossjohn, J. Structural bases of T cell antigen receptor recognition in celiac disease. Curr. Opin. Struct. Biol. 2022, 74, 102349.
- Klöck, C.; Diraimondo, T.R.; Khosla, C. Role of transglutaminase 2 in celiac disease pathogenesis. Semin. Immunopathol. 2012, 34, 513–522.
- Sollid, L.M.; Molberg, Ø.; McAdam, S.; Lundin, K.E.A. Auto-antibodies in celiac disease: Tissue transglutaminase–guilt by association? Gut 1997, 41, 851–852.
- Lexhaller, B.; Ludwig, C.; Scherf, K.A. Identification of Isopeptides Between Human Tissue Transglutaminase and Wheat, Rye, and Barley Gluten Peptides. Sci. Rep. 2020, 10, 7426.
- Du Pré, M.F.; Blazevski, J.; Dewan, A.E.; Stamnaes, J.; Kanduri, C.; Sandve, G.K.; Johannesen, M.K.; Lindstad, C.B.; Hnida, K.; Fugger, L.; et al. B cell tolerance and antibody production to the celiac disease autoantigen transglutaminase 2. J. Exp. Med. 2020, 217, e20190860.
- Stamnaes, J.R.; du Pré, M.F.; Chen, X.; Sollid, L.M. Enhanced B-cell receptor recognition of the autoantigen transglutaminase 2 by efficient catalytic self-multimerization. PLoS ONE 2015, 10, e0134922.
- Alaedini, A.; Green, P.H. Auto-antibodies in celiac disease. Autoimmunity 2008, 41, 19–26.
- Iversen, R.; du Pré, M.F.; Di Niro, R.; Sollid, L.M. Igs as substrates for transglutaminase 2: Implications for autoantibody production in celiac disease. J. Immunol. 2015, 195, 5159–5168.
- Iversen, R.; Roy, B.; Stamnaes, J.; Høydahl, L.S.; Hnida, K.; Neumann, R.S.; Korponay-Szabó, I.R.; Lundin, K.E.A.; Sollid, L.M. Efficient T cell-B cell collaboration guides autoantibody epitope bias and onset of celiac disease. Proc. Natl. Acad. Sci. USA 2019, 116, 15134–15139.
- Petersen, J.; Ciacchi, L.; Tran, M.T.; Loh, K.L.; Kooy-Winkelaar, Y.; Croft, N.P.; Hardy, M.Y.; Chen, Z.; McCluskey, J.; Anderson, R.P.; et al. T cell receptor cross-reactivity between gliadin and bacterial peptides in celiac disease. Nat. Struct. Mol. Biol. 2020, 27, 49–61.

